

Figure3
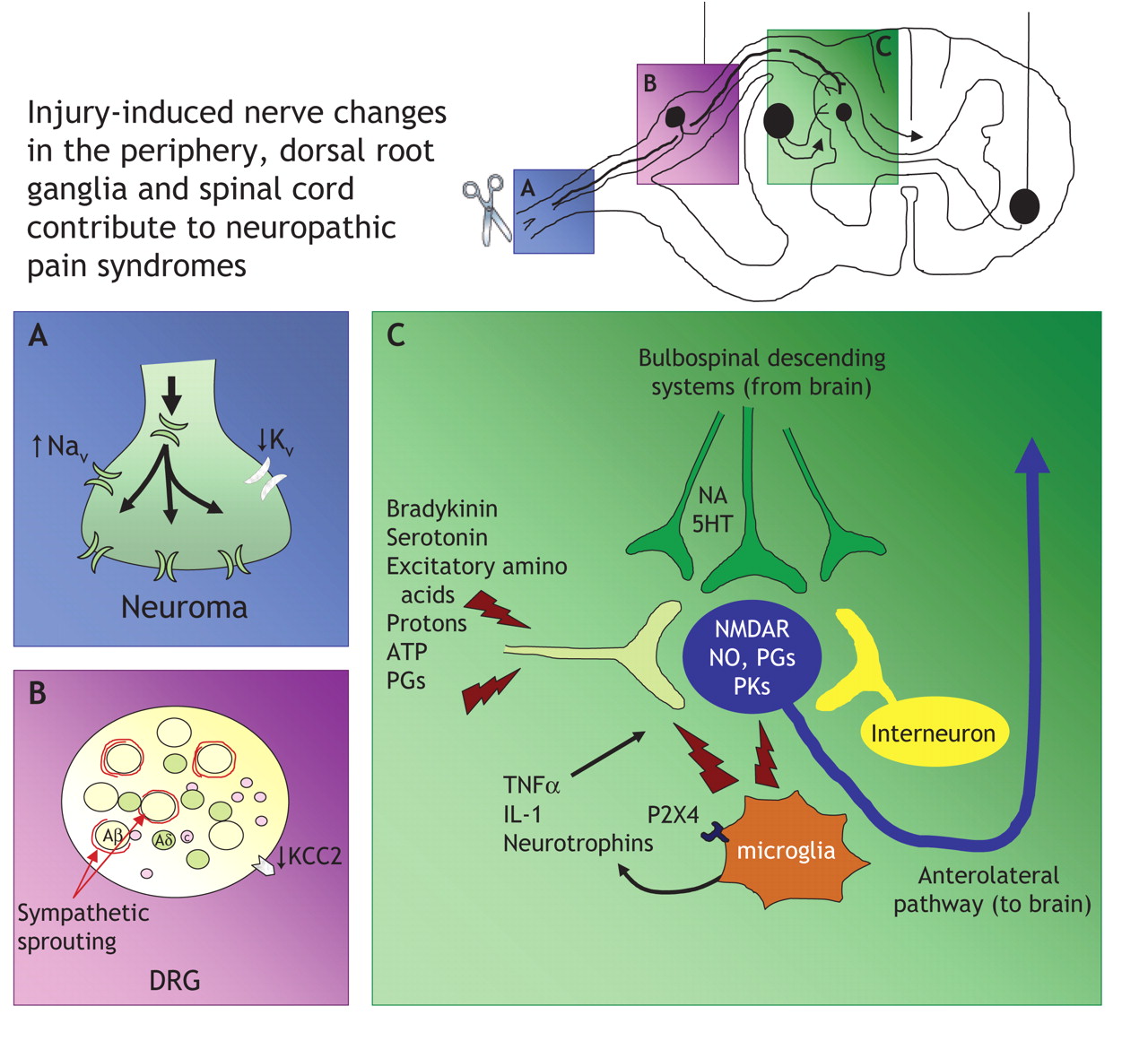
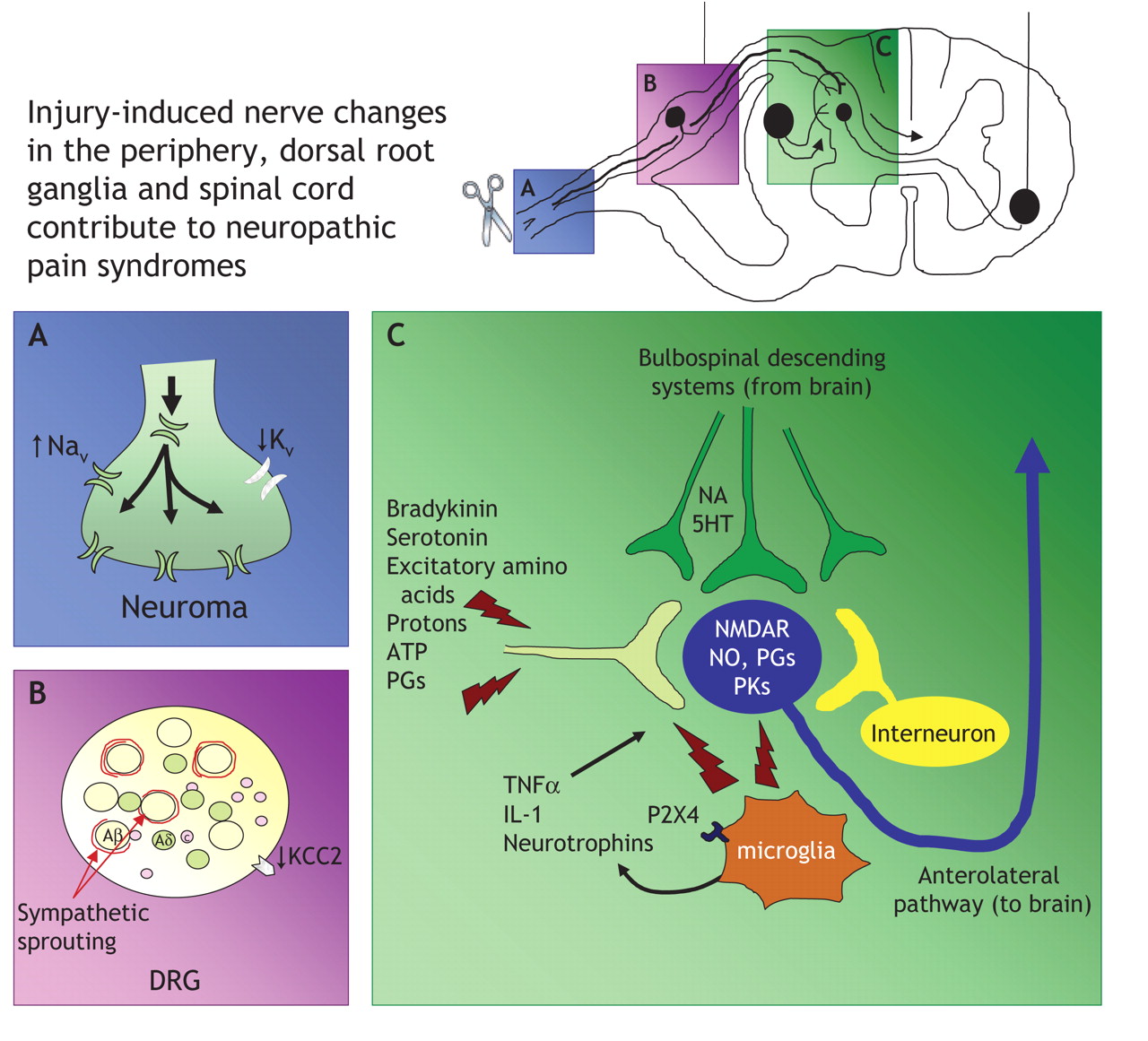
Fig. 2: Neuropathic pain arises following nerve injury or dysfunction. A: After nerve damage, transcription and axonal trafficking of sodium channels to the site of injury is increased, with concomitant attenuation of potassium channels. The altered expression of ion channels results in neurons becoming hyperexcitable and generating ectopic activity, which is thought to lead to the genesis of spontaneous and paroxysmal pain. B: At the cell body of primary afferent neurons within the dorsal root ganglia (DRG), sympathetic neuronal sprouting occurs and may account for sympathetically maintained pain. C: Peripheral nerve injury causes a multitude of changes in gene transcription and activation of various kinases and proteins, including enhanced N-methyl-D-aspartate (NMDA) receptor activity. However, nerve injury also elicits hypertrophy and activation of glial cells, including microglia within the grey matter of the spinal cord. Microglia express P2X4 purinergic receptors, allowing them to be activated by adenosine triphosphate (ATP). Following activation, microglia release various pronociceptive cytokines, such as interleukin-1 (IL-1), tumour necrosis factor alpha (TNF-α) and neurotrophins, including brain-derived neurotrophic factor, which in turn exacerbates nociceptive transmission and contributes to the sensitization and maintenance of neuropathic pain. Note: Aβ = A beta neuron, Aδ = A delta neuron, C = C nociceptor, 5HT = serotonin, KCC2 = chloride transporter, NA = noradrenaline, Nav = sodium channel, NO = nitric oxide, Kv = potassium channel, PGs = prostaglandins, PKs = protein kinases, P2X4 = purinergic receptor.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.↵
- 10.
- 11.
- 12.
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.
- 24.
- 25.
- 26.↵
- 27.↵
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.
- 128.
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.
- 135.↵
- 136.↵
- 137.↵
- 138.↵
In this issue

{kind=link}
Article tools






Related Articles
Cited By...
- Role of peripheral nerve stimulation in treating chronic neuropathic pain: an international focused survey of pain medicine experts
- Etiology and Pharmacology of Neuropathic Pain
- Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement
- Pathophysiology, assessment, and management of pain in critically ill adults
- Management of chronic neuropathic pain: a protocol for a multiple treatment comparison meta-analysis of randomised controlled trials
- Diversification and Specialization of Touch Receptors in Skin
- Relationship of Axonal Voltage-gated Sodium Channel 1.8 (NaV1.8) mRNA Accumulation to Sciatic Nerve Injury-induced Painful Neuropathy in Rats
- Review series: The cell biology of touch
- Pharmacokinetic-Pharmacodynamic Analysis of the Static Allodynia Response to Pregabalin and Sildenafil in a Rat Model of Neuropathic Pain
- P2X4 receptors in activated C8-B4 cells of cerebellar microglial origin
- The Pain Protective Haplotype: Introducing the Modern Genetic Test