


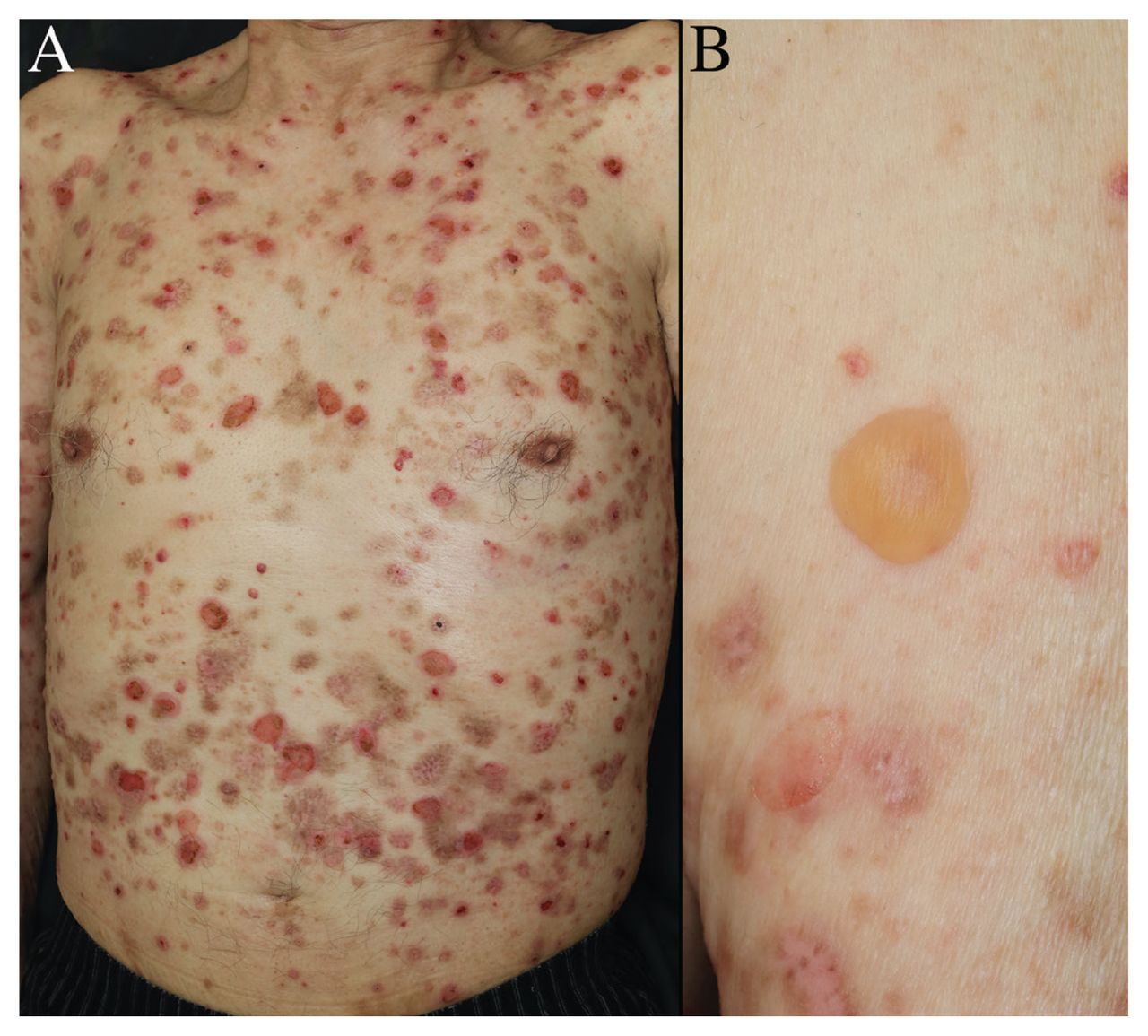
Un homme de 86 ans qui présentait depuis 7 mois des cloques prurigineuses étendues a été adressé à notre clinique de dermatologie par son médecin de famille. L’examen physique a révélé la présence de cloques tendues sur le torse et les membres, ainsi que des zones d’érosion et des croûtes, là où les cloques s’étaient rompues (figure 1). Nous n’avons observé aucune lésion aux muqueuses. Trois ans auparavant, le patient avait commencé à prendre de la téniligliptine pour le traitement du diabète de type 2.

Photographies d’un homme de 86 ans atteint de pemphigoïde bulleuse. (A) Lésions bulleuses, ainsi qu’érosion et croûtes, là où des cloques se sont rompues, au niveau du torse. (B) Cloques tendues sans base inflammatoire sur le bras gauche du patient.
Les analyses sanguines ont montré une numération leucocytaire à 6900/μL (normale 3300–8600), dont 8 % d’éosinophiles (normale 0 %–8 %). Les résultats de la recherche d’anticorps anti-BP180 (domaine NC16A) et anti-BP230 de la pemphigoïde bulleuse étaient négatifs. Une biopsie cutanée a montré la formation de cloques sous-épidermiques (annexe 1, figure 1, accessible en anglais au www.cmaj.ca/lookup/doi/10.1503/cmaj.211933/tab-related-content), et l’immunofluorescence directe a montré un mode de coloration linéaire avec des immunoglobulines G dans la membrane basale. Nous avons diagnostiqué une pemphigoïde bulleuse associée à l’utilisation d’un inhibiteur de la dipeptidyl peptidase-4 (DPP-4). Nous avons cessé la ténéligliptine et entrepris un traitement par de la prednisolone orale, à raison de 40 mg/j pendant 2 semaines, suivi d’un sevrage par paliers hebdomadaires, ce qui a mené à la résolution complète de l’éruption cutanée en l’espace de 2 semaines.
La pemphigoïde bulleuse est une maladie vésiculeuse autoimmune caractérisée par un prurit intense, des cloques tendues et un érythème œdémateux1. Souvent utilisés pour traiter le diabète, les inhibiteurs de la DPP-4 sont associés à la pemphigoïde bulleuse non inflammatoire selon une incidence de 0,42 par 1000 années-personne1–3. Les facteurs de risque rapportés à l’égard de la maladie sont sexe masculin, race blanche et utilisation de vildagliptine ou de linagliptine1,2. La pemphigoïde bulleuse commence habituellement quelques mois à quelques années après l’instauration d’un traitement par inhibiteur de la DPP-43. Contrairement à la pemphigoïde bulleuse inflammatoire, la pemphigoïde bulleuse associée aux inhibiteurs de la DPP-4 a tendance à ne pas s’accompagner d’urticaire ou d’érythème grave à la base des lésions, et les patients sont souvent séronégatifs à l’égard du domaine NC16A de l’antigène BP180, comme chez notre patient3,4. L’arrêt de l’inhibiteur de la DPP-4 entraîne une résolution complète des symptômes chez environ le tiers des patients2. Les patients qui demeurent symptomatiques devraient être orientés en dermatologie pour un traitement par corticostéroïdes ou tétracyclines.
Les images cliniques sont choisies pour leur caractère particulièrement intéressant, classique ou impressionnant. Toute soumission d’image de haute résolution claire et bien identifiée doit être accompagnée d’une légende aux fins de publication. On demande aussi une brève explication (300 mots maximum) de la portée éducative des images, et des références minimales. Le consentement écrit du patient au regard de la publication doit être obtenu avant la soumission.
Remerciements
Les auteurs remercient James R. Valera pour son aide à la révision du manuscrit.
Footnotes
Intérêts concurrents: Aucun déclaré.
Cet article a été révisé par des pairs.
Les auteurs ont obtenu le consentement du patient.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
In this issue

{kind=link}
Article extras
Article tools






Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles

