


A 66-year-old man was referred to an urgent eye clinic after one week of experiencing painless proptosis and epiphora (abnormal overflow of tears down the cheek) in his right eye. The intraocular pressure was normal in both eyes. The patient had normal visual acuity but had right-gaze diplopia. No lymphadenopathy was detected on physical examination. The patient reported no systemic symptoms such as fevers, night sweats and weight loss. He had a history of non–ST-segment elevation myocardial infarction.
Computed tomography (CT) of the patient’s head showed a homogenous enhancing mass 3.7 × 1.7 cm arising from the right lacrimal gland and displacing the lateral rectus muscle (Figure 1). Pathologic analysis of an incisional biopsy specimen showed a dense lymphoplasmacytic infiltrate with storiform fibrosis and occasional eosinophils. Immunohistochemical analysis showed an increased number of immunoglobulin G4 (IgG4)–positive plasma cells (104 per high-power field) constituting 75% of total plasma cells. There was no evidence of obliterative phlebitis, and no granulomas or malignant cells were seen. Flow cytometry performed on the biopsy tissue yielded negative results (Figure 2). Results of tests for rheumatoid factor, angiotensin-converting enzyme (ACE), antinuclear antibodies and antineutrophil cytoplasmic antibodies were negative. Serum calcium and C-reactive protein levels, and liver and renal function test results were within normal limits. Serum immunoglobulin subclass analysis revealed an increased IgG4 level of 3.50 (normal 0.01–1.40) g/L. Based on the overall clinical features, the biopsy results and the elevated serum IgG4 level, a diagnosis of IgG4-related disease was made.

Enhanced computed tomography scan showing homogenous enhancing mass 3.7 × 1.7 cm centred in the right lacrimal gland (red arrow) in a 66-year-old man with painless subacute proptosis.

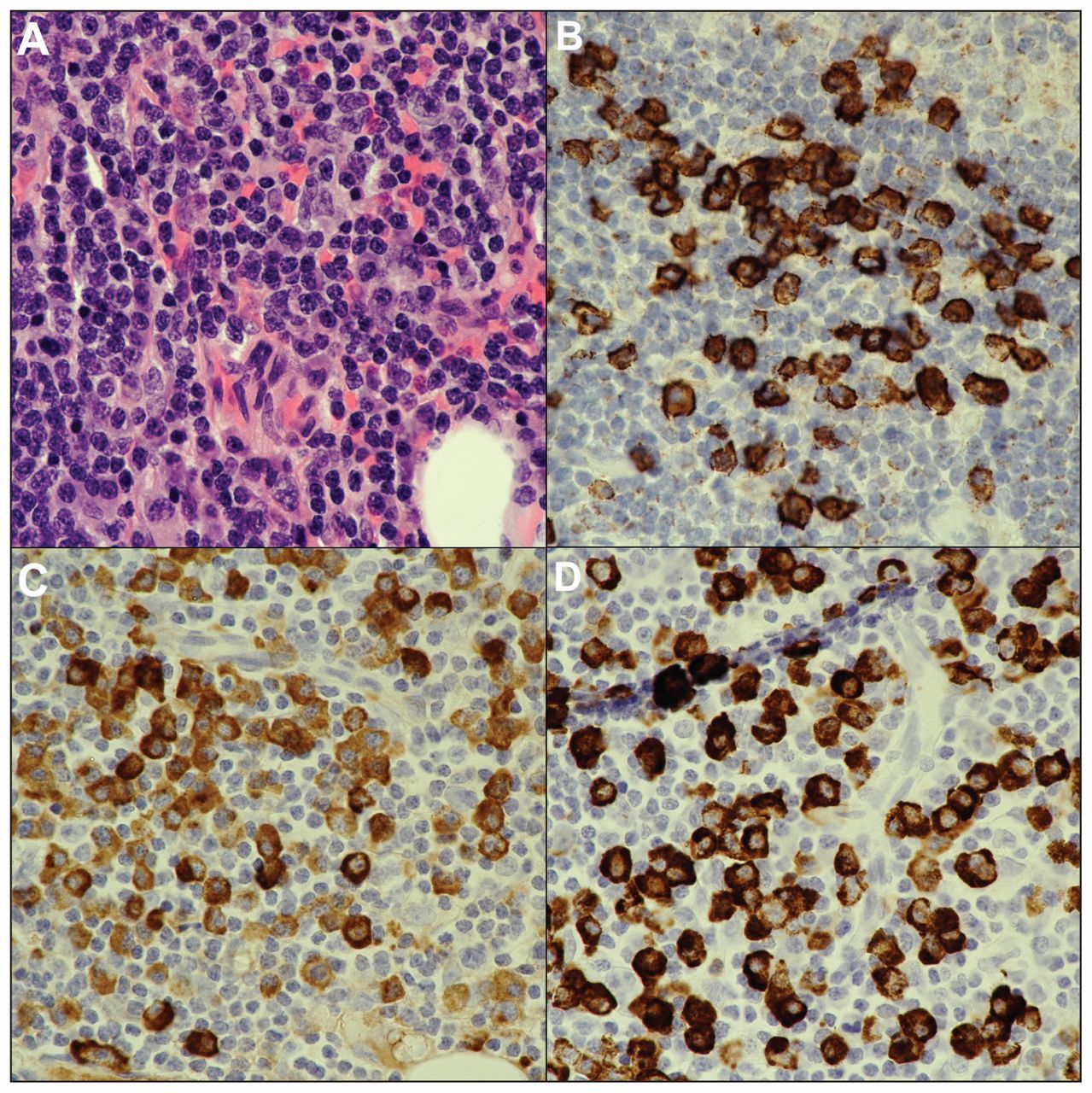
Biopsy of the lacrimal gland showing histologic and immunohistochemical features compatible with immunoglobulin G4 (IgG4)–related disease: (A) dense lymphoplasmacytic infiltrate with mild background fibrosis (hematoxyilin and eosin stain, original magnification × 60); (B) CD138 immunostain showing that the inflammatory cells are plasma cells; (C) IgG immunostain, which stains all IgG classes of plasma cells; and (D) IgG4 immunostain showing that most (about 75%) of the IgG-positive plasma cells belong to the IgG4 subclass.
The patient was prescribed prednisone 50 mg/d orally and followed monthly. His diplopia and proptosis decreased over six months. Follow-up CT of the orbits at six months showed substantial reduction in the size of the mass. The patient did not have clinical evidence of further organ or systemic involvement. A CT scan of the abdomen and pelvis at six months did not suggest systemic involvement. The prednisone dose was tapered over three months, with resolution of symptoms. At one-year follow-up, the patient had no relapse or symptoms suggesting other organ involvement.
Discussion
Immunoglobulin G4–related disease is a recently described systemic fibroinflammatory disease characterized by organ enlargement and fibrosis, an elevated serum IgG4 level and characteristic histopathologic features. Its discovery linked many single-organ diseases, including autoimmune pancreatitis and idiopathic salivary gland fibrosis, under a single disease spectrum by a common pathology. Since the description of the disease-defining IgG4-positive plasma cell infiltrate in 2003, IgG4-related disease has been reported in nearly every organ. Thus, physicians from any specialty may encounter the disease.
The disease should be considered in patients with organ enlargement or dysfunction of unknown cause, especially when multiple systems are involved. Diagnosis requires exclusion of mimics, including malignant disease, autoimmune disorders and atypical infections. A definitive diagnosis is crucial for proper treatment and prognosis, and it should be sought in patients with clinical features suggestive of IgG4-related disease.
History
The discovery of IgG4-related disease began with a landmark series of 20 patients with autoimmune pancreatitis in which elevated serum IgG4 levels were detected in a subset of patients with diffuse pancreatic enlargement.1 Subsequently, IgG4-positive plasma cell infiltrate was identified in pancreatic biopsies from six of the patients.2 Biopsy specimens of their bone marrow, salivary glands and retroperitoneal tissue also contained elevated IgG4-positive plasma cell counts.2 The authors proposed that autoimmune pancreatitis was one manifestation of a systemic process with a distinct pathology uniting organ-specific diseases to the same underlying process.2 Since this discovery, many diseases have been reclassified as organ-specific manifestations of systemic IgG4-related disease.
Epidemiology
The epidemiology of IgG4-related disease is unknown, in part because diagnostic criteria have only recently been standardized,3 the symptoms vary widely and many physicians are unfamiliar with the disease. A widely cited survey of Japanese inpatients estimated the prevalence to be 0.28–1.18 per 100 000, with an average age of patients of 58 years and a slight male predominance.4 Similar findings were reported by a small French registry that identified men in their sixth decade as having the highest prevalence of IgG4-related disease.5
Pathophysiology
The disease’s pathophysiology is not understood. The physiologic role of IgG4 in the immune system has not been elucidated.6 End-organ damage in IgG4-related disease occurs due to infiltration of tissues by lymphocytes, which leads to organ enlargement, reactive inflammation and fibrosis.7 The initial trigger for plasma cell infiltration may be an infectious agent, autoimmune dysregulation or an abnormality of IgG4 itself.6
Clinical manifestations
The pancreas and salivary glands are the most commonly affected organs in patients presenting with IgG4-related disease.8 A systematic review identified the most common symptom as mass effect of the enlarging organ (e.g., patients with IgG4-related pancreatitis presenting with jaundice secondary to enlargement of the pancreatic head).8 Often, the disease is discovered incidentally as a mass on imaging studies performed for other indications.8 Rarely, patients present with subacute organ failure.8 Pathologic studies have identified clinically silent IgG4-positive lymphoplasmacytic infiltrate in tissue samples from patients with symptomatic IgG4-related disease in another organ, which suggests that subclinical IgG4-related disease exists.2,7,8 Lymphadenopathy is commonly found but is not usually a presenting feature.5 Examples of IgG4-related disease exist in virtually every organ, including the kidney (tubulointerstitial nephritis), hepatobiliary tract (sclerosing cholangitis), retroperitoneum (retroperitoneal fibrosis), aorta (aortitis) and thyroid (Riedel thyroiditis).6,8
A retrospective histopathologic review of 65 biopsies performed because of orbital enlargement indicated that IgG4-related disease may account for 10.7%–27.7% of cases initially diagnosed as idiopathic orbital inflammation or orbital benign lymphoid hyperplasia, which are idiopathic conditions that cause nonneoplastic enlargement of orbital tissue.9 Orbital IgG4-related disease is the fifth most common organ-specific manifestation, most often presenting with painless proptosis and diplopia without systemic illness.6,9
Diagnostic criteria
Organ-specific diagnostic criteria for IgG4-related disease have been published.3 Diagnosis requires a compatible clinical history and histopathologic findings and the exclusion of a broad differential diagnosis, and it is supported by an elevated serum IgG4 level.7 Initial evaluation of suspected cases should include serum IgG4 measurement, testing to exclude alternative disorders and tissue biopsy. Referral to a rheumatologist or organ-specific specialist may be necessary for a definitive diagnosis.
Evaluation
Immunoglobulin G4–related disease should be suspected in patients who present with organ enlargement, fibrosis or dysfunction of unknown cause.7 Careful attention should be given to a history of previously unrecognized manifestations of IgG4-related disease.7 Investigations for IgG4-related disease should be considered in patients who have common clinical manifestations of IgG4-related disease, including pancreatitis, sclerosing cholangitis, inflammatory pseudotumour, salivary gland enlargement, retroperitoneal fibrosis and lymphadenopathy of unknown cause.8
An elevated serum IgG4 level is supportive of, but not required for, the diagnosis. Many disorders, including lymphoproliferative diseases and Sjögren syndrome, are associated with elevated serum IgG4 levels.7,10 A retrospective case series comparing 132 patients with pathologic features and a clinical course compatible with a diagnosis of IgG4-related disease and patients with non–IgG4-related inflammatory disorders showed a sensitivity of 97.0% and specificity of 79.6% when the serum IgG4 level was greater than 1.35 g/L.10 Without compatible clinical and histopathologic features, an elevated serum IgG4 level alone is nonspecific.
Tissue biopsy with immunohistochemical staining for IgG4 is an important component in the investigation of IgG4-related disease and is often required to exclude competing diagnoses (Box 1).7 The number of IgG4-positive plasma cells must be elevated, with organ-specific thresholds for diagnosis (typically > 100 per high-power field).3 In addition, the proportion of IgG4-positive plasma cells must be greater than 40% of total plasma cells.3 Further, there must be an increased number of lymphocytes and evidence of fibrosis. Obliterative phlebitis is characteristic but is not a mandatory finding and is often absent in patients with lacrimal gland involvement.3 Importantly, compatible histopathologic features alone are not sufficient to exclude certain competing diagnoses such as lymphoma or Castleman disease.
Diagnostic features of immunoglobulin G4 (IgG4)–related disease3
Required features
Elevated IgG4-positive plasma cell count in biopsy specimen (typically > 100 per high-power field)
Proportion of IgG4-positive plasma cells ≥ 40% of total plasma cells in the biopsy specimen
Increased lymphocyte count in biopsy specimen
Evidence of fibrosis
Exclusion of alternative disorders
Suggestive features
Evidence of obliterative phlebitis in biopsy specimen
Elevated serum IgG4 level
History of other organ involvement
A critical step in the diagnosis of IgG4-related disease is the exclusion of alternative disorders. Clinical, radiologic and pathologic mimics of IgG4-related disease include antineutrophil cytoplasmic antibody–associated vasculitides, Sjögren syndrome, rheumatoid arthritis, pernicious anemia, low-grade B-cell lymphoma, sarcoidosis or other granulomatous disease, primary retroperitoneal fibrosis and malignant disease (Box 2).3 Features of these disorders should be sought by detailed history, physical examination and directed investigations.7 A history of symptoms of unrecognized systemic IgG4-related disease is suggestive. Tests for antinuclear antibodies, antineutrophil cytoplasmic antibodies, rheumatoid factor, urinalysis, serum creatinine, serum calcium with albumin, and complete blood count should be performed, as well as additional investigations suggested by the patient’s history or findings on physical examination.3 Careful histologic review should be performed by a pathologist experienced with IgG4-related disease to exclude these alternative disorders.7
| Differential diagnosis | Differentiating features |
|---|---|
| Immune-mediated | |
| Sjögren syndrome | Anti–SS-A (anti-Ro) and anti–SS-B (anti-La) antibodies |
| ANCA-associated vasculitides | ANCA, ANA, histopathology |
| Granulomatosis with polyangiitis | |
| Eosinophilic granulomatosis with polyangiitis | |
| Microscopic polyangiitis | |
| Giant cell arteritis | Histopathology |
| Rheumatoid arthritis | Rheumatoid factor, anti-CCP antibody |
| Primary sclerosing cholangitis | History of inflammatory bowel disease, histopathology |
| Pernicious anemia | Intrinsic factor antibody |
| Neoplastic | |
| Lymphoproliferative malignant disease | Immunohistochemistry |
| Adenocarcinoma | Histopathology |
| Squamous cell carcinoma | Histopathology |
| Castleman disease | Hyper-IL-6 syndrome (anemia, elevated C-reactive protein level) |
| Other | |
| Idiopathic retroperitoneal fibrosis | Histopathology |
| Sarcoidosis | Serum calcium, histopathology |
Note: ANA = antinuclear antibody, ANCA = antineutrophil cytoplasmic antibody, CCP = cyclic citrullinated peptide, IL-6 = interleukin-6.
Management and prognosis
The cornerstone of treatment for IgG4-related disease is immunosuppression.7 The natural history of the disease is varied; some patients go into remission without treatment, whereas others have rapidly progressive disease. Treatment is started in symptomatic and asymptomatic patients with critical organ involvement to prevent progression to permanent organ fibrosis.7 Conservative management may be considered in asymptomatic patients with noncritical disease such as lymphadenopathy.7
Small series suggest that more than 90% of patients improve with moderate- to high-dose prednisone treatment; however, relapse has been reported in 20%–70% of patients during glucocorticoid tapering.3,5 Azathioprine, methotrexate, rituximab and cyclophosphamide are recommended steroid-sparing agents; of these, rituximab appears to be superior in inducing remission.5,7 Maintenance therapy may be indicated in patients with a high likelihood of relapse (i.e., those with biliary disease, multiorgan involvement, a very high serum IgG4 level or previous relapse7) or when recurrence may lead to organ failure. However, the optimal agent for maintenance of remission is unknown.7 The long-term prognosis of IgG4-related disease is unknown, and patients with the disease require long-term follow-up.
Key pointsImmunoglobulin G4–related disease is a systemic disease that links a spectrum of disorders once thought to be organ specific by a characteristic histopathologic lesion.
Clinical presentation varies by the affected organ(s) and is usually related to mass effect due to organ enlargement or subacute failure secondary to fibrosis.
Physicians in any specialty may encounter IgG4-related disease whether suspected clinically or detected incidentally on imaging or biopsy performed for other indications.
Diagnosis relies on characteristic biopsy features and a compatible clinical history, is suggested by an elevated serum IgG4 level and requires exclusion of alternative disorders.
The primary treatment of IgG4-related disease is immunosuppression with glucocorticoids.
The section Cases presents brief case reports that convey clear, practical lessons. Preference is given to common presentations of important rare conditions, and important unusual presentations of common problems. Articles start with a case presentation (500 words maximum), and a discussion of the underlying condition follows (1000 words maximum). Visual elements (e.g., tables of the differential diagnosis, clinical features or diagnostic approach) are encouraged. Consent from patients for publication of their story is a necessity. See information for authors at www.cmaj.ca.
Acknowledgement
The authors thank Dr. Gary Morris for assisting with the editing of the manuscript.
Footnotes
Competing interests: None declared.
This article has been peer reviewed.
The authors have obtained patient consent.
Contributors: Shannon Ruzycki performed the literature review, and drafted the manuscript and revisions. Margaret Kelly made substantial contributions to the literature review, intellectual content of the manuscript and revisions of the manuscript. Jay Patel identified the case patient, contributed Figure 2 and contributed substantially to the manuscript and its revisions. All of the authors approved the version to be published and agreed to act as guarantors of the work.
In this issue

{kind=link}
{kind=link}
Article tools



Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

