


Abstract
Background: Inflammatory bowel disease is the result of both genes and environment. Canadian First Nations people, despite living in a region with a high prevalence of inflammatory bowel disease, are relatively protected from this disease. We aimed to compare the carriage of genetic variants associated with inflammatory bowel disease in healthy First Nations and white people.
Methods: DNA was extracted from the venous blood of healthy First Nations (n = 340) and white (n = 285) participants from Manitoba. Genotyping was performed for 69 single nucleotide polymorphisms (SNPs) with known or suspected associations with inflammatory bowel disease. We compared the genotypes between groups by logistic regression, adjusting for multiple testing. We calculated a risk score for the NOD2 gene by adding the number of risk alleles at three important NOD2 SNPs (G908R, R702W and 3020insC).
Results: We found genetic variation between white and First Nations participants at 45 of 69 SNPs. Notably, carriage of the ATG16L1 T300A mutation was lower in First Nations participants (p = 4.1 × 10−30). Cumulative carriage of important NOD2 variants was significantly lower among First Nations participants (3.9% v. 15.2%; p < 0.0001 for risk score) than among white participants. Risk variants in IL23R (p = 0.014) and IL12B (p = 1.2 × 10−16), among others, were more prevalent among First Nations participants than among white participants.
Interpretation: The low prevalence of variants associated with bacterial processing and handling in First Nations people may explain their relative protection from inflammatory bowel disease. Increased carriage of a number of risk variants, for example in the interleukin-23/Th17 pathway, is especially intriguing given their importance in other inflammatory diseases of high incidence in First Nations populations.
Inflammatory bowel disease is a chronic immune mediated condition of the gut; its major forms include Crohn disease and ulcerative colitis.1 In people who are genetically predisposed, inflammatory bowel disease is thought to be the result of a loss of tolerance to the gut microflora and consequent aberrant inflammation.
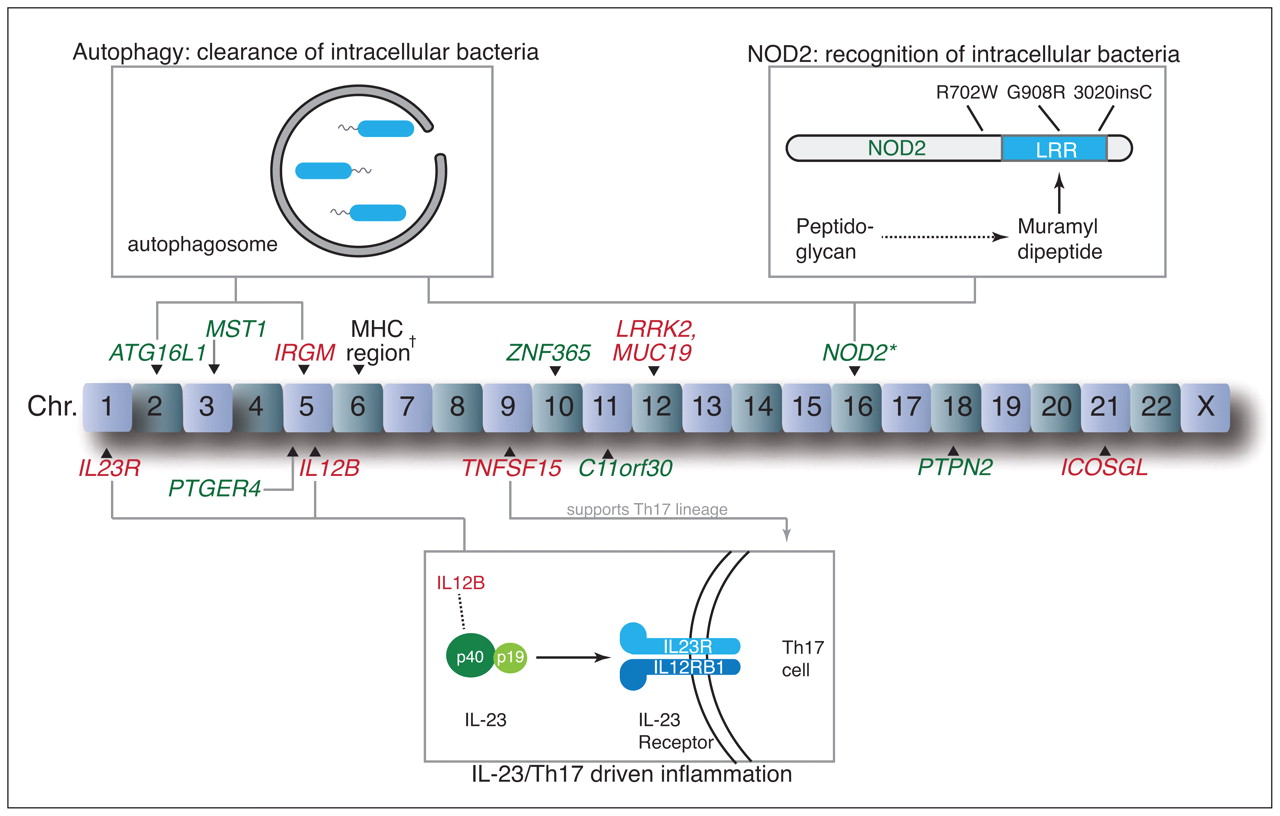
In the past decade, gene association studies have advanced our understanding of the pathogenesis of inflammatory bowel disease (Figure 1).2–10 The first susceptibility gene discovered for Crohn disease, NOD2, encodes a cytoplasmic bacterial pattern-recognition receptor that senses a product of peptidoglycan breakdown, muramyl dipeptide, via its leucine-rich repeat region.2,3,11 Three polymorphisms in or near this region (3020insC, G908R and R702W) collectively encompass 81% of the variants in NOD2 associated with Crohn disease.12 ATG16L1, a gene involved in the intracellular pathogen clearance mechanism known as autophagy, was more recently associated with Crohn disease and further suggests a role for the loss of normal microbial recognition and clearance.8 Proinflammatory immune responses are likely important, given the association of variants in the interleukin-23 pathway, such as IL12B and IL23R, with Crohn disease and ulcerative colitis.4,7 The actions of inter-leukin-23 have largely been linked to Th17 cells, a recently discovered CD4+ subset of T cells thought to contribute to many immune-mediated inflammatory diseases.13

Key loci included in the study and associated biological pathways. The risk variants that we found to be more prevalent among First Nations people are shown in red, and those that we found to be less prevalent are shown in green. *The risk score for NOD2, which combined three important variants (R702W, G908R, 3020insC), was lower in First Nations people. †Prevalence of four variants in the region of the major histocompatibility complex (MHC) differed between groups.
There is heterogeneity in genetic predisposition to inflammatory bowel disease. For example, polymorphisms in NOD2 are prevalent in white but not Asian patients with Crohn disease.14 TNFSF15 has been associated with Crohn disease in both East Asian and white people.10,14,15 TNFSF15 encodes a tumour necrosis factor-like cytokine, TL1A, that drives mucosal inflammation by signalling via the transcription factor NF-κB and can enhance the mucosal function of Th1 and Th17 cells.16
Despite living in an area with a high prevalence of inflammatory bowel disease, Canadian First Nations people appear to be relatively protected. The term First Nations generally refers to members of a band within the meaning of Canada’s Indian Act, encompassing a large proportion of Canada’s indigenous population. Studies undertaken in Manitoba in the 1990s found significantly lower prevalence rates of Crohn disease (16/100 000 people) and ulcerative colitis (56/100 000 people) among First Nations people compared with non-First Nations people (209/100 000 people for Crohn disease and 176/100 000 people for ulcerative colitis).17,18 Nonetheless, this protection from inflammatory bowel disease does not appear to extend to other immune-mediated inflammatory diseases. In particular, many indigenous populations in North America have a high prevalence of rheumatoid arthritis, systemic lupus erythematosus and ankylosing spondylitis (the latter which paradoxically often co-exists with inflammatory bowel disease in other populations).19 Predisposition to rheumatoid arthritis is thought in part to be secondary to the high prevalence of HLA-DRB1 (and thus the shared epitope) in some First Nations people.20 However, studies of the genetics of inflammatory bowel disease in First Nations people are lacking.
In this study, we explored the prevalence of single nucleotide polymorphisms (SNPs) associated with inflammatory bowel disease in healthy First Nations and white people from Manitoba. We hypothesized that First Nations people have a low prevalence of SNPs associated with inflammatory bowel disease, which possibly contributes to their protection against this disease.
Methods
Participants
We recruited healthy First Nations (n = 340) and white (n = 285) people from Manitoba. We excluded people if they or any first-degree relative had inflammatory bowel disease, rheumatoid arthritis or another chronic immune-mediated inflammatory disease. We included First Nations people who had at least three grandparents of First Nations origin. We included white people for whom all grandparents were white; the specific country of origin was not requested.
We recruited healthy white and First Nations people by advertisements posted in the University of Manitoba’s Health Science Centre specialty clinics and in the clinics of participating First Nations communities. The participating communities were Ste Theresa Point, an isolated northern rural community with no road access and an on-reserve population of 2632, and Norway House Cree Nation, a remote semi-isolated northern rural community accessible by road with an on-reserve population of 4071.21 The participants were all unrelated; however, spouses were invited to enrol.
Ethics approval was granted by the University of Manitoba’s research ethics board, and we obtained informed consent from each participant. Samples and data were anonymized. Consistent with the guidelines by the Canadian Institutes of Health Research for conducting research involving Canada’s indigenous population, we established an advisory committee (Autoimmune Diseases in First Nations Advisory Committee) to provide oversight, and we entered into research agreements with the two First Nations communities.
DNA processing
We chose 69 SNPs largely based on previous associations with Crohn disease or ulcerative colitis. The list of SNPs is available in Appendix 1 (available at www.cmaj.ca/lookup/suppl/doi:10.1503/cmaj.110613/-/DC1). We included SNPs associated with inflammatory bowel disease in Asian populations because of the hypothesis that North America became populated as a result of a number of migrations across the land bridge between Eastern Siberia and modern Alaska during the Pleistocene Epoch.22 For example, we included a number of SNPs from a Japanese ulcerative colitis gene association study.9
DNA was extracted from the venous blood of participants and stored at 4 °C. Sixty-eight SNPs were analyzed using the Sequenom iPLEX Gold Assay platform (Génome Québec, Montréal, Quebec). The NOD2 3020insC (rs2066847) SNP was genotyped using TaqMan (The Centre for Applied Genomics, Hospital for Sick Children, Toronto, Ontario). The Sequenom genotyping was unsuccessful for 11 First Nations and 28 white samples.
Statistical analysis
We explored for associations between SNPs and ethnic background using logistic regression. We assumed an additive genetic model. We treated the white group as the control group; thus, odds ratios are shown in relation to the white group. P values are shown with Bonferroni correction for multiple testing (adjusted for 69 SNPs). We also tested for Hardy–Weinberg equilibrium (Appendix 2, available at www.cmaj.ca/lookup/suppl/doi:10.1503/cmaj.110613/-/DC1). For important NOD2 variants (3020insC [rs2066847], R702W [rs2066844] and G908R [rs2066845]), we created a risk score to increase power because the allele frequencies were low in both populations. The number of risk alleles at these three SNPs were summated and compared by Wilcoxon rank sum test. We considered the results to be significant if the corrected p value was less than or equal to 0.05. The number of participants provided power of at least 80% to detect a genetic relative risk of 2.0 at p = 0.001, assuming a minor allele frequency of 0.1.
We tested for linkage disequilibrium using the Broad Institute’s (Boston, Massachusetts) SNAP Version 2.2 and HapMap Release 22. The resulting correlation coefficient (r2) is a measure of linkage disequilibrium between two loci, with a range from 0 to 1.
Results
The characteristics of the First Nations (n = 340) and white (n = 285) participants are shown in Table 1. In total, 336 (98.8%) First Nations participants had four grandparents who were of First Nations ancestry. Of the 69 SNPs tested, 45 were significantly different between First Nations and white participants (Appendix 1). Of these, 28 SNPs were at loci confirmed to be associated with inflammatory bowel disease (Table 2; NOD2 SNPs are also shown). Table 3 summarizes the putative biological relevance of these loci. After correction for multiple testing (data not shown), we found that all SNPs were in Hardy–Weinberg equilibrium (Appendix 2). The allele frequencies among the white participants were generally similar to those described in HapMap (Appendix 2).
Demographics of study participants
Loci associated with inflammatory bowel disease that were significantly different between First Nations and white participants
Summary of loci associated with inflammatory bowel disease that were significantly different between First Nations and white participants and the putative biological relevance of each locus
The prevalence of the T300A risk variant of ATG16L1 (rs2241880) was significantly lower among First Nations participants (p = 4.1 × 10−30). This was confirmed in two nearby SNPs in close linkage disequilibrium (r2 > 0.75), rs3828309 (p = 3.5 × 10−30) and rs3792106 (p = 5.1 × 10−28). A variant associated with Crohn disease (C) at rs11747270 in IRGM was enriched in the First Nations group (p = 0.01). This variant is in close linkage disequilibrium (r2 = 1) with the IRGM SNP (rs13361189) recently shown to be associated with ulcerative colitis.7
None of three NOD2 variants in or near the leucine-rich repeat region that are associated with Crohn disease (R702W, G908R, 3020insC) were independently significantly different between the First Nations and white groups, probably because the risk variants at these SNPs were less prevalent overall (Table 2). To increase the power, we created a risk score by adding the number of risk alleles for each of these three SNPs. The NOD2 risk score was significantly lower among First Nations participants than among white participants (p < 0.0001, Wilcoxon rank sum test), reflecting lower carriage of any of these three NOD2 mutations in the First Nations participants (3.9%) compared to white participants (15.2%). Two other NOD2 risk variants (rs5743289, rs2076756) were significantly decreased in First Nations (p ≤ 1.4 × 10−8). One exception was an intronic NOD2 SNP (rs10521209) identified in a smaller German Crohn disease study;23 this risk variant was more prevalent in First Nations (p = 4.0 × 10−17).
Inflammatory bowel disease variants in the interleukin-23 pathway were enriched in the First Nations group. The risk allele at rs11465804 (in the interleukin-23 receptor subunit gene IL23R) was more prevalent in the First Nations group (p = 0.014). A risk variant associated with Crohn disease (G at rs10045431) in IL12B, which encodes the p40 subunit shared by interleukin-23 and interleukin-12, was more prevalent in the First Nations group (p = 1.2 × 10−16).
The T allele at rs1800896 in the IL10 promoter, which is associated with lower expression of the regulatory cytokine interleukin-10,24,25 was more prevalent in First Nations individuals (p = 1.5 × 10−19; Appendix 1); however, it is not in close linkage disequilibrium (r2 = 0.19) with the confirmed inflammatory bowel disease locus at rs3024505.4,7
We also found differences in genes involved in the immunological synapse and adaptive immune cell activation (Table 2). A variant associated with ulcerative colitis near HLA-B (rs9263739) was less prevalent in First Nations participants (p = 2.7 × 10−11), while the SNP associated with Crohn disease at rs3763313, near HLA-DRB1 among others, was enriched in First Nations participants (p = 4.1 × 10−4). Overall, there were significant differences between groups in inflammatory bowel disease SNPs in the vicinity of the major histocompatibility complex (rs3763313, rs9268480, rs2395185, rs9263739). In addition, two risk variants in TNFSF15 (rs4263839, rs6478108) were enriched in First Nations individuals (p = 2.3 × 10−4), as was one in ICOSGL (rs762421) (p = 7.9 × 10−31).
Of the remaining 17 SNPs that were significantly different between the two groups, 10 were nonreplicated from a Japanese ulcerative colitis gene association study9 (rs12075255, rs885334, rs17351243, rs11583394, rs1862101, rs6746441, rs7591368, rs9553939, rs8004059 and rs379977; Appendix 1). An additional seven SNPs were at loci of interest but were not among the SNPs confirmed to be associated with susceptibility to inflammatory bowel disease in genome-wide association studies, including in the IL10 promoter (rs1800896), IL12A (rs583911), KIF5A (rs1678542), TCF-4 (rs7901695), PTPRS (rs17130), SERPINA1 (rs11832) and rs6128541 (Appendix 1).
Interpretation
We studied the prevalence of SNPs associated with inflammatory bowel disease in First Nations people, using a sample of a Manitoban population in which the low incidence of disease has been previously described.17,18 As expected, given the disparate anthropologic origins of First Nations and white Canadians, there was significant genetic variation at a large number of SNPs. There is a paucity of research chronicling the genetics of First Nations people in Canada, particularly in relation to immune-mediated inflammatory diseases. However, the unique susceptibility and protection patterns in this population for complex diseases, such as inflammatory bowel disease, systemic lupus erythematosus, rheumatoid arthritis and ankylosing spondylitis, make studying the genetics of First Nations people potentially insightful.
We found that First Nations people had a lower prevalence of a number of variants involved in bacterial recognition and autophagy, perturbations in which are thought to be key in the pathogenesis of Crohn disease (Figure 1).26 Especially striking was the low prevalence of ATG16L1 and NOD2 variants. Replacement of the T allele with C at rs2241880 results in the substitution of threonine with alanine at position 300 (T300A) in ATG16L1, which impairs the autophagosomal clearance of intracellular bacteria27 and confers susceptibility to Crohn disease8 and possibly ulcerative colitis.28 NOD2 encodes a cytoplasmic pattern recognition receptor (NOD2) that senses the peptidoglycan breakdown product muramyl dipeptide. In First Nations people, combined carriage was lower of three important NOD2 variants (R702W, G908R, 3020insC) in or near the leucine-rich repeat region responsible for muramyl dipeptide recognition.11 Notably, the autophagy pathway, a process particularly important in the clearance of intracellular bacteria, was recently shown to involve interaction between NOD2 and ATG16L1 proteins.29,30
We found that two variants of TNFSF15, a gene associated with inflammatory bowel disease in East Asian people,14 were enriched in First Nations individuals. This gene’s associations have been thus far specific to inflammatory bowel disease4,7 and ankylosing spondylitis.31 Its product TL1A enhances Th17 function,16 and it may thus work together with variants in IL23R and IL12B to augment this common inflammatory pathway (Figure 1).13 IL23R variants are also associated with anyklosing spondylitis and psoriasis.32 We postulate that risk variants in the interleukin-23/Th17 axis may predispose First Nations individuals to a number of immune-mediated inflammatory diseases, while an absence of variants in bacterial handling and processing pathways prevents development specifically of inflammatory bowel disease.
Although it is tempting to focus on the analysis of genes for which putative pathogenic mechanisms have been shown, we also found differences between the two populations at a large number of loci whose functions are less understood (Table 2). This highlights the complexity of the pathogenesis of inflammatory bowel disease. Future research elucidating the mechanisms at these loci may shed light on their role in First Nations populations.
Limitations
There are many questions unanswered by our study. For example, although First Nations and white Canadians both live in Canada, there are clearly potential environmental differences (e.g., rural versus urban living, diet, rates of diarrheal illness and Helicobacter pylori infection).33,34 Surely some environmental factors contribute to the unique susceptibility patterns of First Nations people toward immune-mediated inflammatory diseases. However, the low incidence of inflammatory bowel disease among First Nations people makes it difficult to study the environmental and genetic factors affecting susceptibility within this population.
The functional significance of many SNPs included in this study is unknown and, in some cases, may reflect linkage disequilibrium with a causal variant in the population in which they were initially studied. This limits the ability to make firm conclusions about the contribution of known inflammatory bowel disease loci to the risk of disease in First Nations people. The use of convenience sampling may limit the ability to generalize our study’s findings.
Conclusion
We found substantial genetic variation between First Nations and white people at loci associated with inflammatory bowel disease. A paucity of risk variants in bacterial sensing and processing pathways and enrichment of variants in the interleukin-23/Th17 axis may reflect the protection and susceptibility pattern of First Nations people toward inflammatory bowel disease and other inflammatory diseases. However, heterogeneity in the relative prevalence of different variants underlines the complexity of inflammatory bowel disease and the unequal contribution of variants to disease pathogenesis.
Footnotes
Competing interests: Charles N. Bernstein is on the advisory board of Abbott Canada, Janssen Canada and Shire Canada. He has served as a consultant for AstraZeneca Canada and has provided expert testimony for Ranbaxy Pharmaceuticals, Barr Pharmaceuticals, Cardinal Health and Mylan. He holds grants from Abbott Canada, Prometheus Laboratories and Aptalis and has received payment for lectures from Abbott Canada, Shire Canada and Merck Canada. Hani El-Gabalawy is on the advisory board for Roche, Bristol-Myers Squibb and Abbott. He holds grants from Roche, AstraZeneca, Abbott and Schering-Plough. He has given pharma-subsidized presentations at academic institutions and has received payment for the development of educational materials from Abbott and has received travel expenses for pharma-related talks. Mark Silverberg holds grants from the National Institute of Diabetes and Digestive and Kidney Diseases and the Crohn’s and Colitis Foundation of Canada. He is a member of the advisory board of and has served as a consultant for Abbott, Janssen and Prometheus Laboratories. He holds grants and has received payment for lectures from these companies. No competing interests declared for all other authors.
This article has been peer reviewed.
Contributors: Travis B. Murdoch, Charles N. Bernstein, Hani El-Gabalawy, Brenda Elias, Joanne M. Stempak, Saad Pathan and Mark S. Silverberg were involved in the study conception and design. Charles N. Bernstein, Hani El-Gabalawy, Michael Sargent, Joanne M. Stempak and Mark S. Silverberg were involved in the acquisition of data. Travis B. Murdoch, Charles N. Bernstein, Hani El-Gabalawy, Wei Xu and Mark S. Silverberg were involved in the analysis and interpretation of the data. Travis B. Murdoch wrote the manuscript, which was critically revised by all other authors. Charles N. Bernstein, Michael Sargent and Mark S. Silverberg provided technical and material support. Charles N. Bernstein, Hani El-Gabalawy and Mark S. Silverberg supervised the study. All of the authors approved the final version of the article submitted for publication.
Funding: This work was funded by a Canadian Institutes for Health Research (CIHR) team grant on Arthritis and Inflammatory Bowel Disease. Dr. Bernstein is supported in part by the Bingham Chair in Gastroenterology. Dr. Silverberg is supported in part by the Gale and Graham Wright Research Chair in Digestive Diseases. Dr. Elias is supported in part by a CIHR New Investigator award. The study sponsors had no role in the design of the study, the collection, analysis or interpretation of data, the writing of the report or the decision to submit the article for publication.
References
In this issue

{kind=link}
Article tools



Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles
Collections

