


- © 2008 Canadian Medical Association
Abstract
Traumatic brain injury is a leading cause of morbidity and death in both industrialized and developing countries. To date, there is no targeted pharmacological treatment that effectively limits the progression of secondary injury. The delayed progression of deterioration of grey and white matter gives hope that a meaningful intervention can be applied in a realistic timeframe following initial trauma. In this review we discuss new insights into the subcellular mechanisms of secondary injury that have highlighted numerous potential targets for intervention.
We provide an overview of traumatic brain injury and, in particular, the mechanisms of secondary injury and emerging novel concepts for future intervention strategies. Understanding these processes in greater detail can identify feasible time frame for treatment and targets for meaningful interventions.
Severe traumatic brain injury continues to be a leading cause of death and morbidity in North America.1–8 The incidence of mild traumatic brain injury is high, and it is a major management problem for clinicians and a considerable source of frustration for patients. The economic and social burden of traumatic brain injury has implications on a global scale, with incidences in developing countries rising as the rate of vehicle use outpaces the development of safety infrastructure.9,10 In addition, traumatic brain injury is now a major focus of casualty care in combat areas, as it is the principal cause of mortality and morbidity especially because of the recent surge in the use of low-cost, yet powerful, explosive devices directed at civilian and military personnel.11
Extensive literature aimed at understanding the tissue, cellular, inflammatory and subcellular processes following traumatic brain injury have proven unequivocally that these pathophysiological events are delayed and progressive in nature. Although the greatest impact on survival and outcome to date may be attributed to systemic and intracranial physiologic management (e.g., fluid resuscitation, intracranial pressure monitoring), future mitigation of the progression of secondary injury will likely be through molecular, gene and pharmacologic interventions. The prospect of gene therapy and pharmacologic treatments require physicians to be familiar with the subcellular mechanisms of brain injury.
Overview of brain injury
A patient is described as having a severe traumatic brain injury if he or she remains in a coma (Glasgow Coma Score of 8 or less) following initial resuscitation. These patients often require mechanical ventilation and invasive monitoring of intracranial pressure. The mortality is between 30%–50% and is higher among older patients.12 Among patients who die from traumatic brain injury, about 90% die within 48 hours of injury, usually because of uncontrolled raised intracranial pressure leading to brain stem herniation and death by neurologic criteria.13 Delayed deaths are caused by either complications in critically ill patients or by decisions to withdraw invasive physiologic support from patients unable to survive without this life support.
Mild traumatic brain injury is much more difficult to define and is likely considerably underdiagnosed. Incidence estimates vary widely, but it may affect 100–600 per 100 000 people annually.14,15 Early identification usually includes a history of direct trauma to the head and brief loss of consciousness. However, mild traumatic brain injury also occurs following acceleration–deceleration forces without direct trauma and often without explicit loss of consciousness. Ongoing symptoms include headache, dizziness and concentration, memory and other neuropsychiatric and cognitive complaints and findings. This is a problem for which there is increasing awareness and identification as well as increasing knowledge of the biological basis of the symptoms.
Approaches to preventing traumatic brain injury can be described as primary, secondary and tertiary. Primary prevention aims to prevent the trauma altogether. Efforts range from changing public policies (e.g., speed limits, helmet use, safety standards, road engineering) to changing public culture (e.g., alcohol abuse, helmet use in recreational activities and preventing sports-related concussions). Secondary prevention is aimed at minimizing the whole biological injury resulting from the trauma. Tertiary prevention refers to maximizing patients' functional abilities and restoring their daily life following an established brain injury. These include various approaches to neuro-rehabilitation as well as symptom management. Tertiary prevention also includes increasing awareness of the consequences of mild traumatic brain injury and understanding the relation between imaging, function and underlying pathology.
Secondary prevention and injury
Secondary prevention comprises minimizing the biological injury arising from the immediate physical trauma and maximizing the biological potential for tertiary prevention. Although concepts of tertiary prevention are now being incorporated into the early management of traumatic brain injury, secondary prevention is the primary focus of prehospital and acute health care delivery. Central to these efforts are 2 assumptions: there are evolving and delayed biological injuries following trauma (“secondary injury”); and interventions directed at secondary injury can make a difference. Certainly the most dramatic evidence for the first concept is a patient who, following traumatic brain injury, “talks and dies” — that is a patient who, at first, is able to verbalize sensibly but subsequently deteriorates, typically because of delayed or evolving intracranial hemorrhage, and dies from rapidly progressive raised intracranial pressure. The early removal of various types of traumatic intracranial hematoma can change the outcome for these patients. Over the last few decades, we have learned much about factors associated with worse outcomes following traumatic brain injury, such as hypotension and hypoxia. It is likely that advances in prehospital care or transport as well as advances in critical care, which have resulted in the reduction of hypoxia and hypotension, may be the reason for the improved outcomes we have witnessed following severe traumatic brain injury.
There is a substantial body of work that has analyzed other systemic and intracranial physiologically targeted interventions that might reduce secondary injury and make a difference in outcomes. These include the analysis of prehospital factors, choice of fluid for resuscitation, blood-pressure management, temperature management, intracranial pressure management, oxygenation parameters and ventilation techniques. These have resulted in the publication of practice guidelines for severe traumatic brain injury.16 Similarly, there is literature that involves the early clinical identification and diagnosis of mild traumatic brain injury and its early and ongoing management. However, much of this work is aimed at the early initiation of strategies for tertiary prevention.
In addition to clinical systemic approaches and those directed by intracranial physiology, clinical studies of interventions targeted at reducing secondary injury arising from discrete subcellular processes (e.g., toxicity due to reactive oxygen species, overstimulation of glutamate receptors, excessive influx of calcium and inflammatory upregulation) have not resulted in a standard drug treatment. This likely reflects both the complexity of the mechanisms of secondary injury as well as issues of drug dosing and timing and the lack of specificity of drug effects.17,18 In the last 10 years, further exploration into the mechanisms underlying the failure of subcellular treatments for secondary injury has revealed a greater than anticipated complexity of molecular processes, but it has also revealed novel targets with higher probabilities of success. Bridging these knowledge gaps is paramount to developing effective future treatments.
Basic biological mechanisms
Although primary injury comprises the initial tear, shear or hemorrhage, the mechanisms of secondary injury can dramatically exacerbate the initial injury. However, the delayed process of secondary injury allows hope for meaningful intervention. Secondary injuries are multiple, parallel, interacting and interdependent cascades of biological reactions caused by primary injury. The major known pathways are summarized in Figure 1 and some clinical manifestations are illustrated in Figure 2.
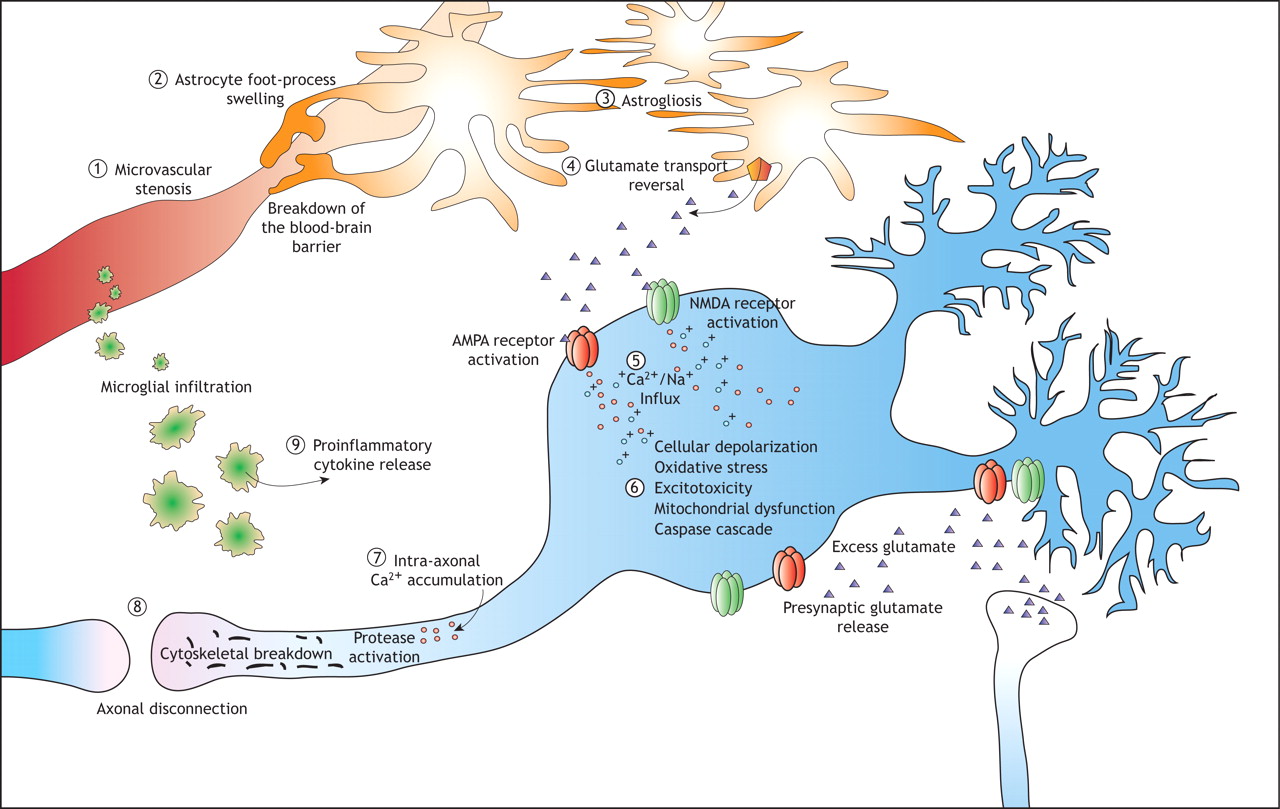
Figure 1: The major pathways associated with the progression of secondary injury after a traumatic brain injury. Microcirculatory derangements involve stenosis (1) and loss of microvasculature, and the blood–brain barrier may break down as a result of astrocyte foot processes swelling (2). Proliferation of astrocytes (“astrogliosis”) (3) is a characteristic of injuries to the central nervous system, and their dysfunction results in a reversal of glutamate uptake (4) and neuronal depolarization through excitotoxic mechanisms. In injuries to white and grey matter, calcium influx (5) is a key initiating event in a molecular cascades resulting in delayed cell death or dysfunction as well as delayed axonal disconnection. In neurons, calcium and zinc influx though channels in the AMPA and NMDA receptors results in excitotoxicity (6), generation of free radicals, mitochondrial dysfunction and postsynaptic receptor modifications. These mechanisms are not ubiquitous in the traumatized brain but are dependent on the subcellular routes of calcium influx and the degree of injury. Calcium influx into axons (7) initiates a series of protein degradation cascades that result in axonal disconnection (8). Inflammatory cells also mediate secondary injury, through the release of proinflammatory cytokines (9) that contribute to the activation of cell-death cascades or postsynaptic receptor modifications.

Figure 2: Computed tomography scans of the brain of a 35-year-old man showing normal anatomy and normal-sized ventricles (left) and a 25-year-old man involved in a motor vehicle crash (right), showing frontal contusions, a depressed skull fracture and compressed ventricles (arrow) from cerebral edema and raised intracranial pressure.
In general, the ongoing sequelae of damage to nervous tissue is perpetuated by the early failure of neuronal energy, glial injury and dysfunction (swelling of astrocytic foot processes, reversal of neurotransmitter reuptake and reactive astrocytosis), inflammation (invasion of the injury site by microglia and release of proinflammatory cytokines), destruction and stenosis of microvasculature, excitotoxicity and aberrant ionic homeostasis in neurons, and progressive white matter deterioration (Box 1).

White matter injury
White matter — the part of the brain that provides long connections between different parts of the grey matter or cortex — exhibits different patterns of deterioration compared with grey matter. Traumatic axonal injury is a common occurrence in both focal and diffuse brain trauma regardless of injury severity.19–22 Traumatic axonal injury has proven to be a reliable predictor of poor survival or poor long-term outcome23–26 yet it is frequently underdiagnosed, particularly in mild traumatic brain injury owing to a lack of tools with sufficient thresholds of detection. Importantly, we now understand that there are mechanisms leading to delayed white matter injury beyond the direct initial consequence of shear forces generated at the moment of initial trauma. Following trauma to the head, especially with rotational forces, it is possible to identify axonal shearing from primary injury. However, a greater number of axons undergo disconnection, or secondary axotomy, at later times.27 Immunohistochemical markers of axonal injury, advanced imaging technologies and serum biomarkers have demonstrated that white matter injury is in fact a progressive and delayed degenerative process. This occurs in both severe and mild traumatic brain injuries.
Abnormal calcium homeostasis is a critical component of the progression of secondary injury in both grey and white matter. In neuronal cell injury, it is associated with excitotoxic cell death, initiation of programmed cell death and postsynaptic receptor modifications. In axonal injury, calcium initiates a cascade of events culminating in axonal disconnection. In both neuronal and axonal injury, calcium overload is linked to early mitochondrial swelling.28,29 Excessive sequestration of calcium by mitochondria causes its membrane depolarization, the opening of membrane permeability transition pores and the release of initiating factors of programmed cell death.30,31 The loss of mitochondrial function not only eliminates calcium buffering capacity but also contributes to the influx of calcium resulting from bioenergetic failure of ATP-dependent ion pumps. For example, cyclosporin A, an immunosuppressant and inhibitor of the mitochondrial membrane-permeability-transition pore, has been shown to reduce both axonal pathology and neuronal cell loss, thus illustrating the importance of this process.32–35
White matter injury is increasingly recognized as central to the impact on the quality of life of patients with either severe or mild traumatic brain injury. After severe injury, damage to the major white matter tracts is devastating and, despite normal intracranial pressure, it can be the determinant of death or persistent disability (Figure 3). Following mild traumatic brain injury, white matter lesions may be the determinant of ongoing symptoms as well as patients' increased susceptibility to future traumatic events.

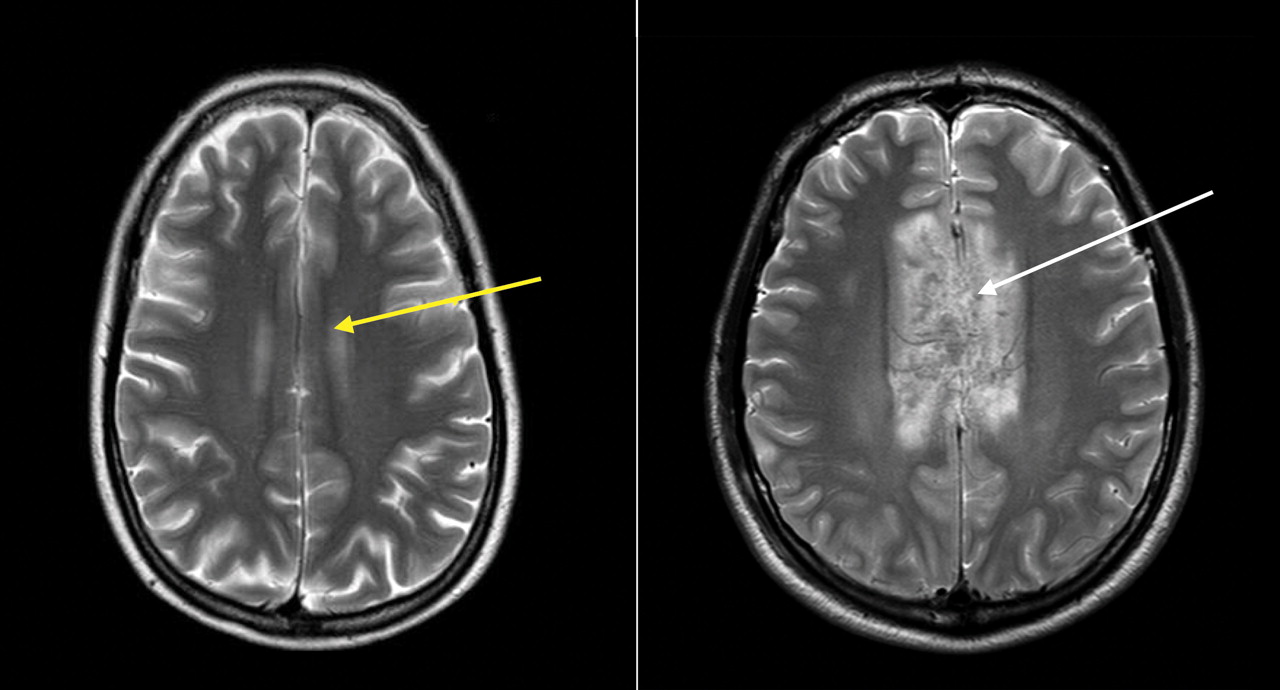
Figure 3: Magnetic resonance images of the brain of a 38-year-old woman (left) and 35-year-old male passenger in a motor vehicle crash (right) with extensive injury to the corpus callosum (a major tract of white matter between the left and right cerebral cortex) (white arrow). The yellow arrow in the left panel shows an area of the corpus callosum with no edema or disruption.
The important secondary injury of white matter involves the axonal membrane becoming “leaky” (a state permitting an influx of extracellular calcium).36–38 As a result of increased calcium concentrations in the axon, enzymes that degrade key structural proteins become activated and destroy proteins responsible for the maintenance of shape and transport in axons. These events cause the accumulation of transported proteins, axonal swelling and, ultimately, disconnection39
Activation of protein-targeting enzymes after trauma can result in the production of distinct protein fragments. The identification of these signature markers through proteomic screening technologies has high diagnostic potential for determining the nature and severity of injury as well as potential therapeutic targets.40
One family of ubiquitous enzymes, calpains, has received considerable attention as key mediators of axonal injury. Given the profound protection conferred by pharmacologic antagonists in animal models, these enzymes may also be potential therapeutic targets for white matter injury.41–43 Under normal physiologic conditions (low intracellular calcium), calpains have a wide range of targets with numerous regulatory functions.44,45 Under pathophysiological conditions, however, calpains target axonal proteins responsible for structure and transport.45–47 A particular axonal protein expressed exclusively in the brain, αII-spectrin, provides an example of how the diagnosis of white matter injury may be aided by proteomic technologies. Calpain-mediated breakdown of αII-spectrin results in the production of 2 specific fragments (150 kDa and 145 kDa).48,49 In contrast, caspase-3 cleavage (typically associated with programmed cell death) of αII-spectrin results in a 150 kDa and a 120 kDa fragment.48 This distinction allows the dominant process to be determined and holds potential for diagnosis and directed drug design.
Several conventional histologic markers used to characterize traumatic injury to axons include the degradation of several intra-axonal cytoskeletal proteins and the accumulation of transported proteins, such as β-amyloid precursor protein.50–52 As an increasing number of markers of white matter injury are elucidated, it is becoming evident that histologic examination of a few select markers does not reflect the full extent of axonal injury because of the diverse spectrum of mechanisms that can occur as a function of injury severity, location and tissue type. A key stumbling block in the development of treatments for white matter injury is identifying the mechanisms that are most damaging or, more realistically, interventions that can be applied within a reasonable time frame.
Although histologic markers can be used to characterize the pathological events that occur in white matter, the effective translation of basic research findings into meaningful clinical therapy ultimately depends on the functional outcome of white matter. In this regard, electrophysiological assessments in experimental laboratories have shed light on interventions that translate to functionally measurable outcomes. These experimental techniques have been used to track the functional deterioration of white matter after trauma, to identify subpopulations of axons that are more vulnerable than others and to evaluate potential therapeutic treatments for the deterioration of white matter.53–56
Studies have revealed that white matter trauma is an active progressive process. Following trauma, white matter exhibits active lesions of inflammation, cytoskeletal breakdown and, finally, axonal disconnection. It is a phenomenon known to occur across a range of injury severities. Further complicating the issue is the finding that all axons are not created equal, with varying susceptibilities dependent on subcellular make-up. We are beginning to recognize that white matter lesions may be responsible for more symptoms than previously thought and that they may be the source of the ongoing neuro-cognitive issues that affect patients following mild traumatic brain injury.
Excitotoxicity
Following traumatic brain injury there are excessive extracellular concentrations of the excitatory neurotransmitter glutamate (reviewed by Van Landeghem and colleagues57 and Yi and Hazell58). Failure of presynaptic membrane-bound ion pumps and calcium-mediated exocytosis is thought to contribute in large part to the initial depolarization-dependent release of glutamate from the neurons.59–61 This excess neurotransmission is thought to contribute to the toxic increase in intracellular calcium concentrations. However, more recent theories suggest that excitotoxicity can occur because of trauma-induced changes to postsynaptic glutamate receptor pharmacology, kinetics and subunit composition, and that calcium-mediated toxicity is highly contingent on the location of the calcium influx.
Glutamate receptors can be broadly categorized as AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors or NMDA (N-methyl-D-aspartic acid) receptors based on their responsiveness to chemical agonists. Changes to the function of both receptor subtypes have been identified following modelled traumatic brain injury, ultimately leading to calcium overload and neuronal hyperexcitability.
To study trauma-induced changes at the single-cell level, many laboratories make use of primary cell culture models of tissue strain, mimicking on a cell culture plate the type of axonal and somatic stretching that occurs in the brain because of a rapid acceleration–deceleration injury. The most commonly used method involves culturing neurons on a deformable membrane. This model provides a cellular analogue of the changes to angular momentum that occur during rotational head trauma (e.g., whiplash from a car accident, falls from large distances, athletic-related head injury). The length of time over which tissue strain is produced and its intensity can be varied by the researcher. Subsequently, single-cell electrophysiological characterizations of the cell's responses are measured.
AMPA receptors
A number of groups have identified strain-induced changes to AMPA-receptor transmission. Consistently, traumatic injury to cortical neurons results in an increased current response to AMPA-receptor agonists. Injured neurons display much larger AMPA-receptor ionic currents, potent hyperexcitability, increased free intracellular calcium and susceptibility to otherwise nontoxic concentrations of synthetic glutamate receptor analogues.62–66 In the presence of reduced AMPA-receptor desensitization or overactivity, the neurotoxicity of even a brief period of excess synaptic glutamate after trauma can lead to hyperexcitability, epileptic activity or delayed calcium-dependent cell swelling, damage and death. The intracellular mechanisms that regulate the excitability of AMPA receptors are fairly well established and are currently the targets of many investigations seeking to attenuate the altered function of AMPA receptors following acute injury.
AMPA receptors are composed of a combination of 4 subunits (GluR1–4). Receptors lacking the GluR2 subunit are highly permeable to calcium (reviewed by Isaac and colleagues67 and Chen and colleagues68). This has led to the proposal that a reduction in the GluR2 content of synaptic AMPA receptors might underlie the observed abnormal calcium homeostasis or hyperexcitability in the central nervous system after brain trauma. Interestingly, tumour necrosis factor α, a proinflammatory cytokine known to infiltrate the injury site following traumatic brain injury,69–72 significantly reduces synaptic levels of GluR2 in cultured neurons65,73 and increases surface expression of AMPA receptors lacking GluR2. Coupled with increased synaptic glutamate, this remodelling of the composition of AMPA receptors by tumour necrosis factor α (released from injured glia and inflammatory cells) leads to post-injury calcium overload. This cross-talk between an inflammatory mediator and glutamatergic neurotransmission sheds new light on how delayed excitotoxicity mediated by AMPA receptors might occur.
It is also known that AMPA receptors lacking GluR2 are highly zinc permeable.67,68 Further evidence supporting the hypothesis that there is a reduction of GluR2 following traumatic brain injury is the marked elevation in free ionic zinc in neurons following experimental brain trauma.74–76 Free zinc is taken up by mitochondria in an effort to restore zinc homeostasis but, similar to the effects of mitochondrial calcium uptake, this leads to potent mitochondrial dysfunction, prolonged loss of mitochondrial membrane potential and free radical generation.77,78 This further implicates the loss of GluR2 as an important step in neuronal death and damage.
NMDA receptors
In addition to hyperexcitability mediated by AMPA receptors, numerous studies of traumatic neuronal degeneration have implicated excessive activity of NMDA receptors.79–84 The intracellular coupling of the NMDA receptor to the generation of reactive oxygen and nitrogen species makes it a deadly route of ion flux (particularly calcium) following a glutamatergic spill. For example, the generation of neuronal nitric oxide is highly dependent on the activity of the NMDA receptor. Though nitric oxide (by definition a free radical) reacts slowly with most biological molecules, when combined with other free radicals (e.g., superoxide generated by compromised mitochondria), it is remarkably reactive.85
During periods of excitotoxicity, excessive free calcium is sequestered by the mitochondria in an effort to restore intracellular calcium homeostasis. The elevated calcium level in the mitochondria increases the production of reactive oxygen species, including the superoxide anion. The reaction of mitochondrial-derived superoxide with NMDA-derived nitric oxide produces the highly reactive nitrating species peroxynitrite (an oxidant with activities similar to that of the hydroxyl radical and nitrogen dioxide radical).86 Peroxynitrite, produced in excess following experimental traumatic brain injury,80,82,87 produces nitration of amino acid aromatic rings,85 lipid peroxidation88 and DNA fragmentation85,89 — all of which are rapidly fatal cellular processes.
One of the more promising post-traumatic brain injury therapeutic interventions involves the perturbation of the physical interaction between the NMDA receptor and the generation of neuronal nitric oxide. By severing the interaction between the receptor complex and scaffolding proteins, investigators have successfully attenuated trauma-induced generation of peroxynitrite and nitric oxide in vitro80 and have circumvented ischemic infarction in animal models of stroke.90 Investigators are currently evaluating the efficacy of this approach in whole animal models of brain injury.
Conclusions
Our understanding of traumatic brain injury has evolved considerably in the last decade, owing in part to renewed research efforts following a series of ineffective trials targeting the mechanisms of secondary injury. There are clearly different mechanisms, both temporal and subcellular, that govern the delayed destruction of white and grey matter. However, in both cases, calcium entry is a critical step in initiating the degenerative processes. A comprehensive understanding of the discrete subcellular mechanisms is critical to bridging the knowledge gaps that will allow effective treatment options to be developed. The problems of mild and severe traumatic brain injury will not be addressed without advances in our ability to prevent secondary injury.
The delayed and active molecular processes involved in white matter deterioration and neuronal cell death gives hope that therapeutic treatment options will be developed. Generalized theories of white matter injury have been challenged by studies illustrating highly complex and parallel mechanisms of injury in axonal subpopulations. Theories of excitotoxicity and cell death have evolved from simple concepts about energy failure and non-specific postsynaptic calcium influx to theories that include distinct trauma-induced alterations to the properties of postsynaptic receptor ion channels and intracellular interactions. An increased awareness of the complexity of secondary injury raises the possibility of more specifically targeted therapeutic strategies, many of which are being currently validated in preclinical studies.
There is no question that primary prevention of severe and mild traumatic brain injury is the desired approach to this common and devastating problem. Nevertheless, advances in the prevention of secondary injury are of great importance for cases in which primary prevention has failed. The goal of clinicians and scientists in this field is to identify the sequence of events in secondary injury with the aim of developing targeted, specific interventions to improve the outcomes of severe and mild traumatic brain injury.
Key points
• Severe traumatic brain injury is a leading cause of morbidity and mortality worldwide.
• Abnormal intracellular calcium homeostasis is a central feature of both grey and white matter secondary injury.
• White matter injury is important in the prognosis of both severe and mild injury.
• Recent studies point to trauma-induced changes in neuronal receptor composition that render cells vulnerable to delayed injury.
Footnotes
-
Une version française de ce résumé est disponible à l'adresse www.cmaj.ca/cgi/content/full/178/9/1163/DC1
Contributors: Each of the authors contributed to the conception and design of the review, drafted and revised the manuscript for important intellectual content and approved the final version for publication.
Competing interests: None declared.
REFERENCES
- 1.↵
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.
- 22.↵
- 23.↵
- 24.
- 25.
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.
- 34.
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.
- 52.↵
- 53.↵
- 54.
- 55.
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.↵
- 63.
- 64.
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.
- 71.
- 72.↵
- 73.↵
- 74.↵
- 75.
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.
- 82.↵
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
- Targets of Neuroprotection and Review of Pharmacological Interventions in Traumatic Brain Injury
- Neuroprotective derivatives of tacrine that target NMDA receptor and acetyl cholinesterase - Design, synthesis and biological evaluation
- RNA-seq analysis and compound screening highlight multiple signalling pathways regulating secondary cell death after acute CNS injury in vivo
- Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo
- Cardiolipin-Dependent Mitophagy Guides Outcome after Traumatic Brain Injury
- Battlefield brain: Unexplained symptoms and blast-related mild traumatic brain injury
More in this TOC Section
Similar Articles

