


- © 2005 Canadian Medical Association or its licensors
THE CASE: A 28-year-old First Nations man was transferred from a northern nursing station to the Health Sciences Centre in Winnipeg for investigation of a 1-day history of generalized weakness that began when he woke in the early morning. He had no fever, myalgia, or gastrointestinal, respiratory or cardiovascular symptoms, and no history of recent illness or known toxic ingestion. He did have a 3-year history of bilateral blindness, for which he had never sought medical attention. His family history was significant only for type 2 diabetes mellitus in his father and several maternal and paternal aunts and uncles.
On examination, he was alert, tachycardic (heart rate 140 beats/min) and afebrile. He had flaccid paralysis of his arms and legs (0/5) and could not lift his head off the bed. His wrist and digit strength were preserved (5/5). Deep tendon reflexes could not be elicited. Sensation to light touch and pinprick were normal. He had normal rectal tone. His visual acuity was decreased because of bilateral cataracts, but the cranial nerves were otherwise intact.
The patient had hypokalemia, with a serum potassium level of 1.6 (normal range 3.5–5.3) mmol/L, and his creatine phosphokinase level was slightly elevated at 328 (normal range 52–175) U/L. Results of other initial laboratory investigations were normal, including complete blood count, and renal and liver function tests.
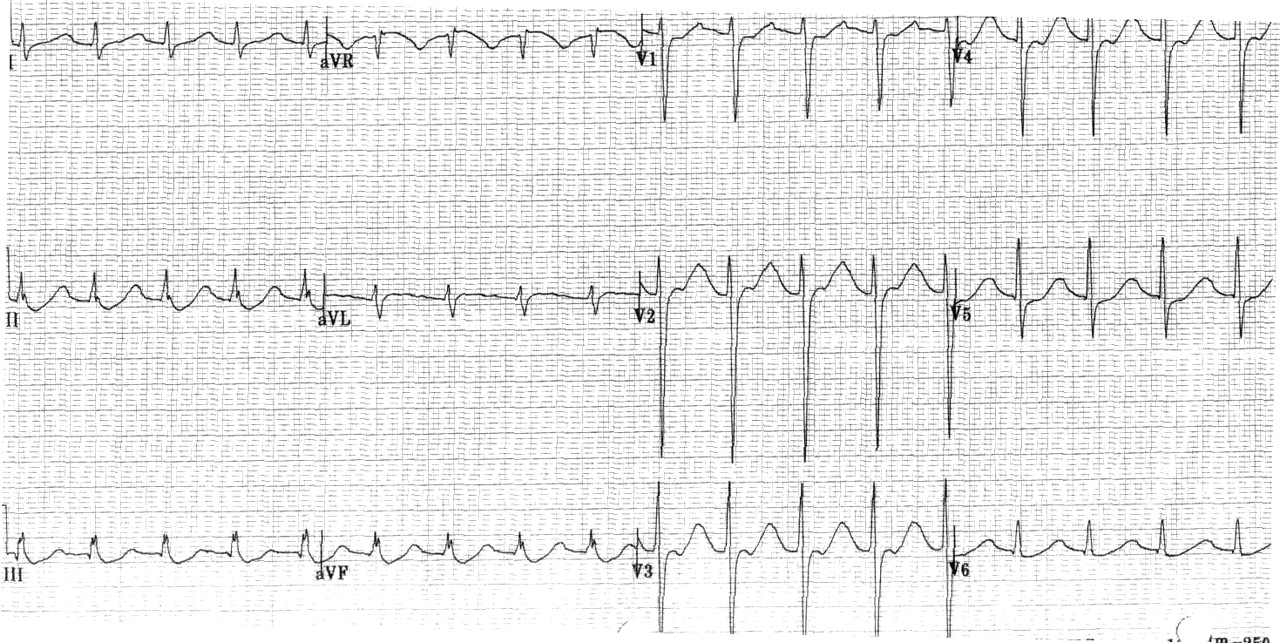
An electrocardiogram revealed sinus tachycardia, a prolonged QT interval corrected to 664 ms, and ST-segment depressions inferiorly in leads II, III and avF (Fig. 1). A head CT revealed chronic bilateral retinal detachment as the cause of the patient's blindness but no intracranial abnormality to explain his weakness.
Twenty-four hours after symptom onset the patient still had hypokalemia (serum potassium level 1.9 mmol/L), despite receiving a total of 200 mEq of potassium orally and 160 mEq parenterally. His distal lower limb strength had improved to 5/5, with some return of strength in his shoulders and hips (2/5). He was still unable to lift his head off the bed. Mild hyperreflexia was now present in his lower limbs.
Dramatically, 2 hours later, the patient was walking independently. His serum potassium level was normal (5.0 mmol/L), as was an electrocardiogram. Potassium replacement therapy was discontinued.
The results of thyroid function testing revealed thyrotoxicosis, with a thyroid stimulating hormone level of < 0.01 (normal range 0.4–4.2) mU/L, an elevated free thyroxine level of 57 (normal range 9.7–25.7) pmol/L and a free triiodothyronine level of 18 (normal range 3.7–6.9) pmol/L. The patient agreed to treatment with propranolol followed by the addition of tapazole, but he declined radioactive iodine. At 10-month follow-up no paralysis had recurred.
Thyrotoxic periodic paralysis (TPP) is a disorder characterized by thyrotoxicosis, hypokalemia and predominantly proximal lower limb paralysis. Despite the female predominance of hyperthyroidism, TPP occurs most frequently in males in a ratio of 20:1, and 90% of patients are of Asian descent.1
Prodromal symptoms may consist of muscle stiffness or cramping. Sensation, as well as bulbar and respiratory muscle strength, remains intact. Reflexes may be decreased or absent.
Attacks may be precipitated by a high carbohydrate meal (secondary to insulin secretion) or physical exertion. In one series, 84% of attacks occurred between 1 and 6 am.2 Episodes are acute in onset and last 1–96 hours. As in this patient, symptoms and signs of hyperthyroidism may be subtle at initial presentation.
Electrocardiographic findings such as ST-segment depression with T-wave flattening and the presence of U waves are typical of hypokalemia. Findings supportive of a diagnosis of TPP are sinus tachycardia, elevated QRS voltage and first-degree AV block (sensitivity 97%, specificity 65%).3
The exact pathophysiology of TPP is unknown. Thyroid hormone itself has a direct effect in stimulating the sodium– potassium– a denosinetriphophatase (Na–K–ATPase) pump. Pump sensitivity to adrenergic stimulation may be higher in patients with TPP than in those with hyperthyroidism alone, resulting in intracellular potassium shift and subsequent hypokalemia. Some human leukocyte antigen subtypes (DRw8, A2BW22, AW19B17, B5, BW46) and genetic mutations (KCNE3) have been associated with TPP.4
Potassium replacement therapy is the mainstay of treatment for TPP. It should be administered carefully because of the risk of rebound hyperkalemia and resulting cardiac complications. Recent literature has advocated instead for the use of β-blockers to reverse the adrenergic overstimulation of the Na–K–ATPase pump by elevated thyroid hormone levels. Evidence suggests that propranolol monotherapy reduces the duration of the TPP event and is associated with a lower risk of rebound hyperkalemia than potassium replacement therapy.5 Although β-blockers may be used in the acute setting, definitive management requires correction of the hyperthyroid state.
The differential diagnosis of acute weakness is summarized in Box 1. Broad categories to consider are electrolyte abnormalities, muscular disorders and central nervous system disorders at spinal cord, synapse and peripheral nerve levels. Specific causes of hypokalemic paralysis are summarized in Box 2.


In summary, TPP should be suspected in patients presenting with acute proximal lower limb weakness. Management should include necessary resuscitative efforts, followed by β-blocker therapy and careful administration of potassium replacement therapy. Further attacks may be avoided by treatment of the hyperthyroid state.
In this issue

{kind=link}
{kind=link}
{kind=link}
Article tools



Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

