


A previously healthy 48-year-old Chinese woman presented with a one-week history of redness, blurred vision and discomfort in both eyes. She also had a frontal headache, and tinnitus and hearing loss in both ears. There was no history of head injury.
The patient was alert and oriented. She had nuchal rigidity and a low-grade fever (temperature 37.6°C). Examination of the cranial nerves gave normal results, and there were no cerebellar or long tract motor or sensory signs. There was no rash or lymphadenopathy.
The patient’s visual acuity, according to a Snellen chart, was 20/30 in each eye. Both pupils had normal direct and consensual light reflexes; visual field, colour vision and the intraocular pressure in each eye were within normal limits. Slit-lamp biomicroscopy of the anterior ocular segment showed mild conjunctival injection and anterior uveitis without hypopyon. Proptosis and orbital bruit were absent. There were no external changes in the ears, and the tympanic membranes in both ears were normal.
A complete blood count and erythrocyte sedimentation rate were within normal limits. Lumbar puncture showed clear cerebrospinal fluid and an opening pressure of 16 (normal 10–20) cm water. Analysis of the cerebrospinal fluid showed 185 leukocytes/mm3 (100% lymphocytes), a protein concentration of 0.2 (normal 0.1–0.5) g/L and a glucose level of 4.0 (normal 2.2–4.2) mmol/L. No micro-organisms were identified in a Gram stain and fungal smear. Magnetic resonance imaging of the head without contrast showed no abnormalities.
What is the next step?
Which of the following would be the least appropriate next step?
-
Administer broad-spectrum intravenous antibiotics without delay.
-
Administer systemic steroids.
-
Refer the patient to an ophthalmologist for fundal fluorescein angiography.
-
Refer the patient to an otolaryngologist for audiometry.
The answer is (b). Systemic steroids should not be administered before an underlying infection, which could be the cause of this clinical presentation, has been excluded. In a patient with clinical and laboratory features of meningitis, it is important to administer empirical broad-spectrum antibiotics until cerebrospinal microbial cultures return negative results or when clinical judgement supports discontinuing therapy. This patient had clinical features of lymphocytic meningitis and anterior and posterior uveitis, which suggested a uveomeningeal syndrome.
The causes of uveomeningeal syndromes include autoimmune, infectious and neoplastic disorders; therefore, a thorough investigative work-up should be done before systemic steroids are given.
An ophthalmologic evaluation — including stereoscopic examination of the fundus and fundal fluorescein angiography — can help determine the intraocular extent of uveitic involvement and identify the diagnostic fundal and angiographic markers of some syndromes with uveitis. Early assessment of the type, extent and severity of uveitis by an ophthalmologist is important to ensure timely treatment and prevent adverse effects. Although an otolaryngologic evaluation can help identify sensorineural hearing loss associated with some uveomeningeal syndromes, it is not required for the immediate management of this patient. Ear examination and magnetic resonance imaging of the brain showed no abnormalities, so it is unlikely that the acute hearing loss was caused by an emergency condition such as acute infection.
With stereoscopic fundal examination, the ophthalmologist detected multifocal choroiditis and subretinal fluid collection in both eyes. A bullous serous retinal detachment was present in the left eye. The optic discs were normal, and no macular lesions were present (Figure 1).

Figure 1: Fundus montage of both eyes of a 48-year-old woman, showing discrete, well-circumscribed yellowish areas representing foci of choroiditis and focal areas of subretinal fluid. Bullous serous retinal detachment is present in the left eye (arrow).
Intravenous fundal fluorescein angiography, ordered by the ophthalmologist, showed multiple areas of pinpoint hyperfluorescence and pooling of fluorescein within the subretinal fluid collections in the late venous phase (Figure 2). A chest radiograph and results of a purified protein derivative test were negative, and the Venereal Disease Research Laboratories test and treponema pallidum hemagglutination assay gave negative results.
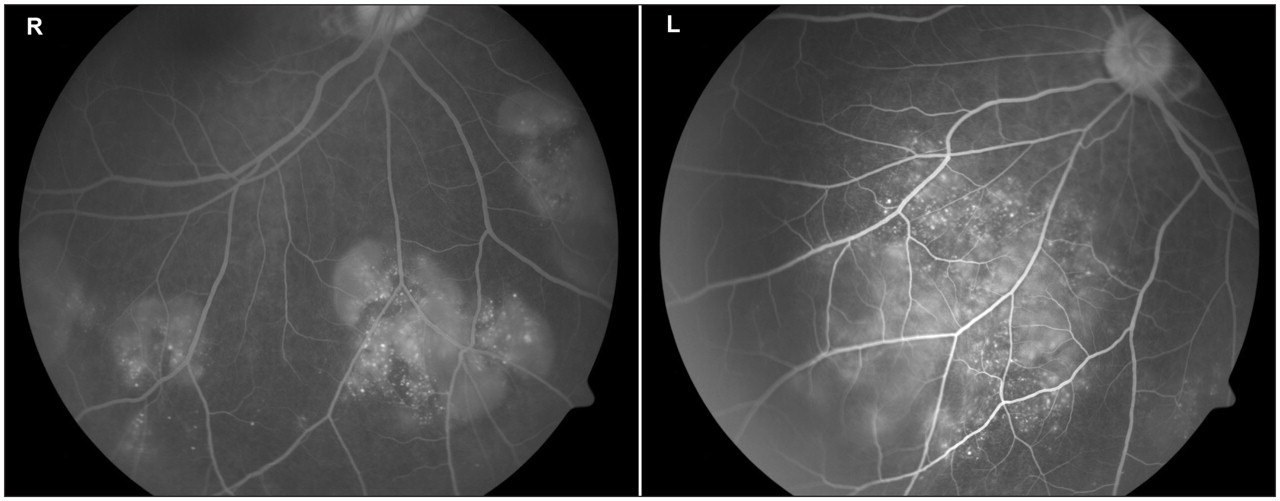
Figure 2: Fundal fluorescein angiography of both eyes in the late venous phase (right eye, inferior view; left eye, inferonasal view) showing multiple areas of pin-point hyperfluorescence, large placoid areas of hyperfluoresence and pooling of fluorescein within the subretinal fluid collections and retinal detachments.
The reviewing otolaryngologist ordered audiometry, which showed mild to moderate bilateral sensorineural hearing loss in the high frequency range (2 to 8 kHz).
What is your diagnosis?
-
Cat-scratch disease
-
Vogt–Koyanagi–Harada syndrome
-
Tuberculosis
-
Sarcoidosis
-
Primary central nervous system lymphoma
Discussion
The answer is (b), Vogt–Koyanagi–Harada syndrome. This patient had characteristic systemic features (meningitis, sensorineural hearing loss, and no previous penetrating ocular trauma, surgery or systemic disease) and ocular features (multifocal choroiditis and subretinal fluid collection) — the diagnostic criteria for this syndrome (Table 1). The absence of dermatologic features supports incomplete Vogt–Koyanagi–Harada syndrome.
Table 1: Causes and types of uveomeningeal syndromes
Referral to an ophthalmologist may be delayed because patients with this syndrome usually present with vague ocular complaints, good visual acuity and obvious meningitis. With Vogt–Koyanagi–Harada syndrome, visual acuity may be preserved if the subretinal fluid collections do not involve the macula. A high index of suspicion is required.
Our patient was treated with 1% prednisolone acetate eye drops every three hours and prednisolone 60 mg daily, administered orally. Both medications were tapered off at four months, after a reduction in uveitis and resolution of the retinal detachments. At six months after presentation, the patient was stable without any recurrence. Her visual acuity is maintained at 20/30 in both eyes. The skin changes characteristic of Vogt–Koyanagi–Harada syndrome did not appear.
Uveomeningeal syndromes (Table 1) manifest with involvement of the eye and central nervous system. Initially, they can be confused with infectious meningitis. Patients present with ocular symptoms of uveitis, such as blurred vision, redness, periocular pain or floaters, and neurologic conditions such as meningitis, encephalitis or cranial neuropathy. Although the term uveitis usually refers to inflammation of the uveal tract, it is now also used to describe retinal inflammation. 1
Uveitis is classified according to the site of the intraocular inflammation into anterior uveitis (iris), intermediate uveitis (pars plana, vitreous and peripheral retina) and posterior uveitis. Posterior uveitis includes papillitis (swelling of the optic disc), choroiditis (discrete yellow subretinal lesions), retinitis (thickening with retinal edema) and vasculitis. The diverse causes of uveomeningeal syndromes are broadly classified into autoimmune, infectious and neoplastic categories.
The approach to a patient presenting with both ocular and neurologic symptoms begins with careful history-taking and systemic and ophthalmic examination, followed by ancillary investigations such as magnetic resonance imaging, analysis of cerebrospinal fluid and fundal angiography. Table 1 shows the infectious, autoimmune and neoplastic differential diagnoses to consider in a patient presenting with a uveomeningeal syndrome. Differentiating systemic, neurologic and ophthalmic features are highlighted in Table 2. 2
Table 2: Differential diagnoses of uveomeningeal syndromes 2
Vogt–Koyanagi–Harada syndrome
Vogt–Koyanagi–Harada syndrome is an idiopathic, multi-system autoimmune disorder characterized by bilateral granulomatous uveitis with neurologic, auditory and dermatologic manifestations. It has been reported that this syndrome accounts for 9.2% of patients in uveitis clinics in Japan; however, this prevalence is likely to be lower in Western populations. 3
The pathophysiology of this disorder involves T-cell-mediated autoimmunity against melanocyte-associated antigens in the choroid, meninges, cochlea and skin. In Japanese studies, the syndrome has been shown to be associated with immunogenetic marker HLA-DR4. 3 Vogt–Koyanagi–Harada syndrome occurs mostly in Native, East Indian, Asian, Middle Eastern and Hispanic people and is uncommon in white and black people. 3 It is more common in women and in patients between 20 and 50 years of age. 3
There is no specific diagnostic test for this syndrome; the diagnosis is based on a combination of clinical features and results of fundus fluorescein angiography and testing of cerebrospinal fluid (Box 1). 4 Sympathetic ophthalmia has an almost identical clinical presentation to Vogt–Koyanagi–Harada syndrome, except for a previous history of penetrating ocular injury or surgery in either eye (Table 1). Bilateral granulomatous panuveitis with multifocal choroiditis, serous retinal detachment, Dalen–Fuchs nodules and sunset-glow fundus (Figure 3) are also seen in Vogt–Koyanagi–Harada syndrome. The syndrome progresses through four stages (Box 2).

Figure 3: Sunset-glow fundus in the right eye with focal areas of hyperpigmentation.

Box 1: Revised diagnostic criteria for Vogt–Koyanagi–Harada syndrome 4

Box 2: Phases of Vogt–Koyanagi–Harada syndrome
The treatment for this condition is early administration of high-dose systemic corticosteroids, which is associated with less intraocular inflammation, decreased frequency of ocular recurrences and dermatologic features, and better visual outcomes. 5–7 Vogt–Koyanagi–Harada syndrome can be complicated by visually significant sequelae such as cataract, glaucoma, subretinal fibrosis and choroidal neovascular membranes. The risk of at least one complication developing is over 50%. 8
Footnotes
-
Previously published at www.cmaj.ca
This article has been peer reviewed.
Competing interests: None declared.
Acknowledgement: The authors thank Dr. Ho Su Ling, National Healthcare Group Eye Institute, Singapore, for providing Figure 3 from her personal collection.
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles
Collections

