


Abstract
Background: In August and September 2002 an outbreak of West Nile virus (WNV) infection occurred in southern Ontario. We encountered a number of seriously ill patients at our hospitals. In this article we document the clinical characteristics of these cases.
Methods: We conducted a retrospective chart review of patients who came to the attention of infectious disease or neurology consultants or the microbiology laboratories at 7 hospitals in the municipalities of Toronto, Peel and Halton, Ont. Patients were included if they had been admitted to hospital or stayed overnight in the emergency department, had serological evidence of WNV infection and had clinical evidence of WNV fever, aseptic meningitis, encephalomyelitis or motor neuronopathy.
Results: In all, 64 patients met the inclusion criteria; 57 had encephalitis or neuromuscular weakness or both, 5 had aseptic meningitis, and 2 had WNV fever. The mean age was 61 years (range 26–87). The patients were predominantly active, middle-aged or elderly people living independently in the community. Seven patients were immunocompromised A febrile prodromal illness preceded the neurological symptoms in almost all cases. The most common neurological abnormality was decreased level of consciousness; this frequently evolved to severe lower motor neuron neuromuscular weakness. Ataxia and swallowing disorders were frequent and important problems. Sixteen patients (25%) required intubation and mechanical ventilation because of a decreased level of consciousness, inability to clear secretions or respiratory muscle weakness; 9 others had disabling muscle weakness of one or more limbs. Ten patients died. The study patients were in hospital a total of 1856 patient-days, including 532 patient-days in an intensive care unit. Only 28% (13/47) of the patients who survived encephalitis or neuromuscular weakness, or both, were discharged home without additional support. Slow turnaround time for serological test results resulted in delayed diagnosis.
Interpretation: The 2002 WNV infection outbreak in Ontario caused serious morbidity and mortality in the subset of patients who had encephalitis or neuromuscular weakness severe enough to require hospital admission.
West Nile virus (WNV) infection has been described in endemic and epidemic forms throughout Africa and the Middle East since the 1930s.1,2,3,4,5,6 It was first documented in North America in 1999, in New York City.7,8 The total number of human cases in North America remained modest until 2002, when a 65-fold increase in the number was documented in the United States.9 In 2001, WNV activity was detected in birds and mosquitoes in 12 health regions in southern Ontario, but no human disease was documented.10 In 2002, WNV infection was detected in Ontario birds collected as early as May 19 in the Peel region.11 Throughout the summer, increasing numbers of WNV-infected mosquitoes were detected, including those of the Culex pipiens species, which feed exclusively on birds, and numerous other mosquito species that have been implicated as secondary bridge vectors believed to have a major role in transmitting the virus to humans.12,13 During August and September 2002 we encountered many patients admitted to hospital with a clinical syndrome compatible with WNV infection. We describe here the clinical characteristics of those cases.
Methods
Information on patients with WNV infection was collected through a collaboration of infectious diseases, microbiology, infection control and neurology staff at 7 Toronto-area hospitals (University Health Network, Mount Sinai Hospital, North York General Hospital, St. Joseph's Health Centre [Toronto], Credit Valley Hospital, Halton Healthcare Services – Oakville Trafalgar Site and Sunnybrook & Women's College Health Sciences Centre). These hospitals accept 42% of acute care admissions in the 3 municipality areas (Toronto, Peel and Halton). Case finding was done through the infectious diseases and neurology consultation services and through hospital microbiology laboratories. Because of publicity before and during the WNV season, WNV serologic testing was included in the diagnostic workup of many patients with meningoencephalitis and peripheral neuropathy. However, active surveillance for human WNV infection was not done in 2002, so the complete extent of WNV activity is unknown.
The definitions used for this study are listed in Appendix 1. Patients were included if they were admitted to hospital or stayed overnight in the emergency department, had fever, had aseptic meningitis, encephalitis or neuromuscular weakness, and had serological evidence of WNV infection (4-fold increase in WNV antibodies assayed by hemagglutination inhibition, or a single titre of 1:320 or greater by hemagglutination inhibition and confirmed by plaque reduction neutralization assay). These inclusion criteria are a modification of the US Centers for Disease Control and Prevention (CDC) criteria14 and of those described by Tardei and colleagues.15 The hemagglutination inhibition and plaque reduction neutralization assays were performed as described previously.16,17 Patients with a history of travel to arboviral endemic areas or of vaccination against yellow fever or Japanese encephalitis were included only if a 4-fold rise in flavivirus antibody levels was documented and the antibodies were confirmed to be WNV-specific by means of plaque reduction neutralization assay.
Using a standardized form, we obtained data by reviewing inpatient hospital charts and clinic charts (for follow-up visits) and by interviewing patients and their families. For calculation of length of hospital stay, data were censored after Dec. 19, 2002. For calculation of time to death, data were not censored after Dec. 19, because 2 of the deaths occurred after that date. The Karnofsky Performance Status Scale18 was used to determine functional status before illness.
Results
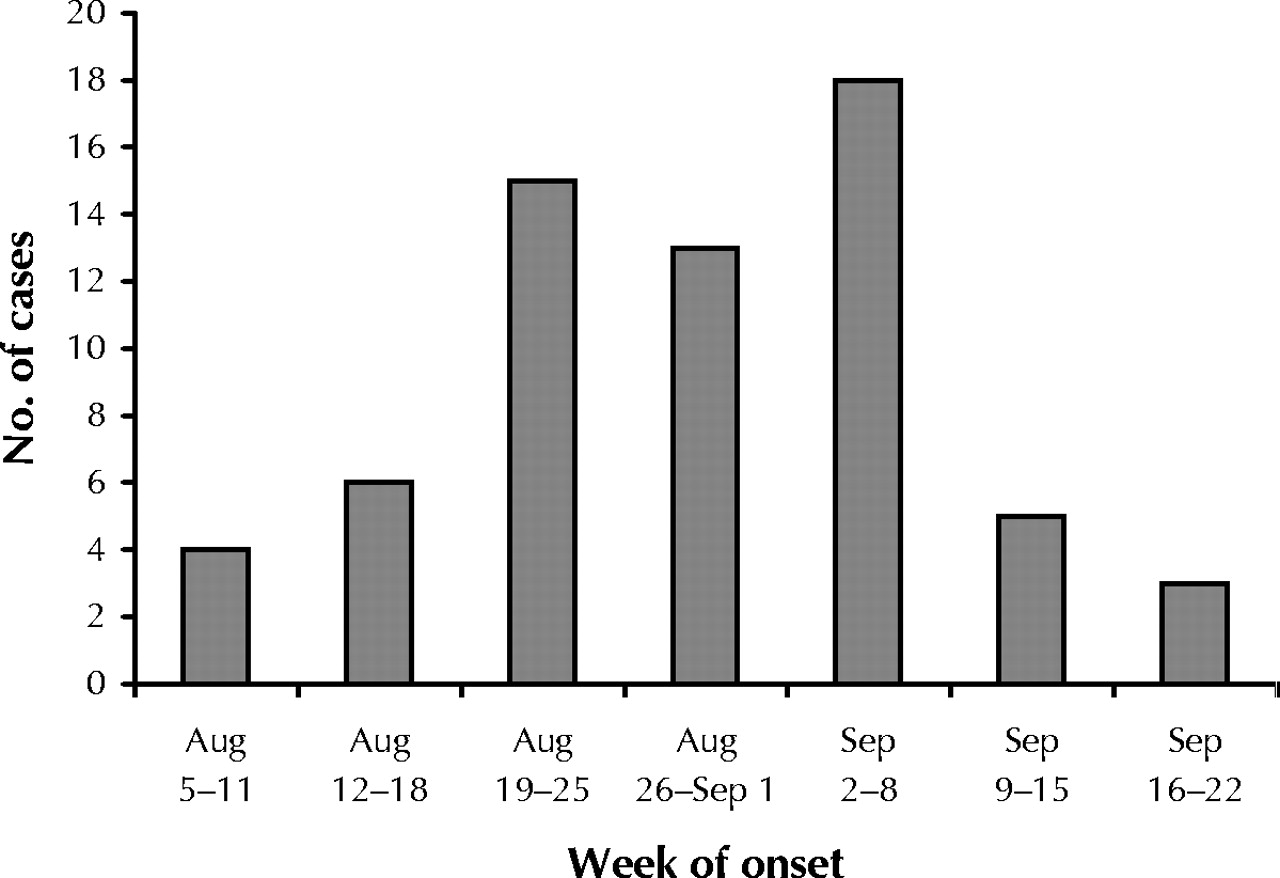
Sixty-four cases of human WNV infection met the inclusion criteria. Illness onset occurred between Aug. 6 and Sept. 20, 2002, and peaked in the first week of September (Fig. 1). Patient characteristics are summarized in Table 1. The mean age was 61 years (median 62.5, range 26–87). Seven patients (11%) were immunocompromised. Thirty-two patients (50%) had significant underlying chronic medical conditions. Despite these underlying illnesses, 44 (72%) of the 61 patients for whom information on functional status was available had a Karnofsky score of 100 before their WNV illness. Only 6 patients (10%) had been unable to function independently before their illness (Karnofsky score ≤ 70), and no one had been a resident in a long-term care facility.
Table 1.
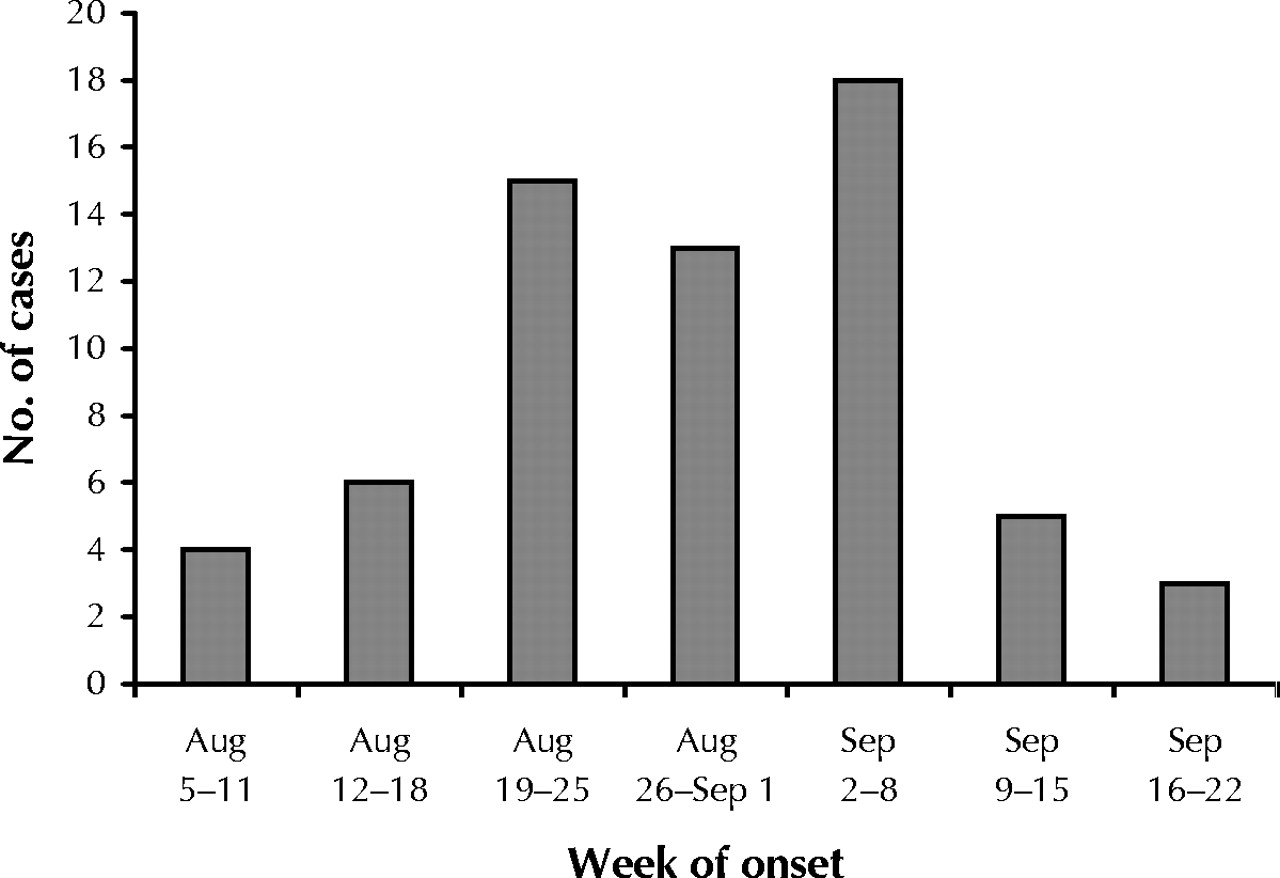
Fig. 1: Number of people with West Nile virus infection admitted to 7 Toronto-area hospitals in 2002, by week of onset.
The symptoms and signs at presentation are listed in Table 2. The fever was usually high (mean peak temperature 39.3°C, median 39.1°C, range 37–40.5°C) and persisted after admission to hospital (mean duration 6 days, range 2–14). Myalgia was noted in 55% of the patients (26/47). Altered mental status and gastrointestinal symptoms were the most common localizing features. Clear evidence of meningeal involvement was often lacking: only 27% of the patients (17/64) had both headache and neck stiffness and 20% (13/64) had neither symptom. In 7 cases, the initial presentation was suggestive of sepsis, with fever, rigors, hypotension, tachycardia and tachypnea. One of these patients required inotropic support in the intensive care unit (ICU) during the first 2 days of admission. An erythematous macular or maculopapular rash was noted in 27% (17/64).
Table 2.
The admitting diagnoses were meningitis or encephalitis, or both, in 24 cases (38%), fever of unknown source in 19 (30%), bacterial infection such as pneumonia or pyelonephritis in 7 (11%), alcohol withdrawal in 3 (5%), stroke in 2 (3%), and myelopathy, multiple sclerosis and transverse myelitis in 1 case each (2%); information on the admitting diagnosis could not be discerned from the medical records in 6 cases (9%).
Neurological manifestations
Five patients had uncomplicated aseptic meningitis and 2 had WNV fever without neurological manifestations. The remaining 57 patients had meningoencephalitis or neuromuscular weakness, or both. Neurological dysfunction usually started several days after the onset of the systemic symptoms (fever, myalgia and gastrointestinal distress) and frequently evolved after admission to hospital. Table 3 presents the frequency of neurological symptoms and signs, and Fig. 2 illustrates the distribution of patients with various combinations of the 3 principal types of neurological manifestations (decreased level of consciousness, brainstem and cerebellar signs, and neuromuscular weakness).
Table 3.

{kind=link}
{kind=link}
Fig. 2: Venn diagram, showing 3 principal types of neurological manifestations of West Nile virus infection: decreased level of consciousness (yellow circle), brainstem and cerebellar signs (pink circle) and neuromuscular weakness (blue circle). Most patients had abnormalities in all 3 areas ( n = 18), decreased level of consciousness alone ( n = 15) or a combination of decreased level of consciousness and brainstem/cerebellar signs ( n = 12).
Encephalitis was noted in 55 patients, 24 of whom also had neuromuscular weakness. Only 2 patients had weakness without signs of encephalitis (Fig. 2). Of the 55 cases of encephalitis, most (48) manifested as decreased level of consciousness (delirium in 21, stupor in 15 and coma in 12). Mental status changes persisted for a mean of 17 (median 10, range 1–67) days. In 15 (27%) of the 55 encephalitis cases, the clinical presentation was limited to a febrile illness with decreased level of consciousness. Other common neurological manifestations included dysphagia (22 patients), cerebellar ataxia (20) and neuromuscular weakness (26) (Table 3, Fig. 2). Less common neurological signs included facial weakness, ophthalmoplegia, dysarthria, nystagmus and myelopathy (Table 3). Cerebellar dysfunction was obvious at presentation in 15 of the 57 patients with neurological manifestations; it developed or became evident several days or weeks into the illness in 5 patients, often when the mental status returned to normal and the patient attempted to walk.
Four patients had severe brainstem disease with loss of brainstem reflexes, labile vital signs and features of “locked-in syndrome.” Two of these patients were immunocompromised (one had undergone a liver transplantation and the other had had lymphoma treated with autologous stem-cell transplantation). In these 2 cases, T2-weighted and FLAIR (fluid-attenuated inversion recovery) MRI scans showed increased signal throughout the brainstem, thalami and vermis of cerebellum. Both patients died, and postmortem examination of one revealed marked inflammation in the grey matter of the thalami, basal ganglia, brainstem and cerebellum. Similar changes were found in the spinal cord grey matter, and there was destruction of anterior horn cells. The peripheral nervous system showed inflammation of the dorsal root ganglia and roots (radiculitis).
Neuromuscular weakness was present in 26 (46%) of the 57 patients with neurological manifestations and was documented days to weeks after the onset of systemic or central nervous system dysfunction. The clinical pattern of weakness consisted of flaccid quadriparesis (17 patients), asymmetric paraparesis (6) and monoparesis (3). Satisfactory electromyography and nerve conduction studies were performed in 14 of the patients with weakness: anterior horn cell or motor axonal dysfunction was found in the majority (10/14). Three patients had mixed axonal degenerating and demyelinating processes, and 1 had a pure demyelinating process. The striking features in the patients with neuromuscular weakness were the extent of flaccid paralysis and the degree of muscle denervation shown on needle electromyography that suggested very pronounced anterior horn cell or motor axonal injury.
Cerebrospinal fluid (CSF) studies were performed in 56 cases: 4 had an elevated protein level, 4 had an elevated leukocyte count, and 48 had both an elevated protein level and pleocytosis. The CSF leukocyte count ranged from 0 to 1179 х 106/L, usually with a lymphocyte predominance (mean 60%, range 3%–100%), although polymorphonuclear leukocytes often predominated early. The mean CSF protein level was 0.9 (range 0.3–2.0) g/L. Six patients had abnormal lymphocytes in the CSF on cytological examination; this finding prompted investigation for lymphoma in 2, with negative results. A third patient, who had undergone lumpectomy and radiotherapy for localized breast cancer, was incorrectly diagnosed as having leptomeningeal carcinomatosis; the abnormal cells seen in her CSF were later shown to be reactive lymphocytes.
CT scans of the brain (in 58 cases) showed no acute abnormalities. MRI of the brain (in 24 cases) showed no acute changes except in the 2 immunocompromised patients described earlier. One other patient had MRI evidence of acute transverse sinus thrombosis.
Acute serologic testing for WNV infection was ordered a median of 2 days after admission (mean 4, range 0–18 days). In 24 cases (38%), WNV antibodies were not detected in the initial (acute) serum specimen. Three of the immunocompromised patients (organ transplant recipient, stem-cell transplant recipient, person with congenital myelodysplasia) had very delayed antibody responses: no response was detected at 25 days (2 patients) and 52 days (1 patient) after the onset of illness. Serum samples from these patients were tested again at 40, 58 and 74 days after illness onset, and high WNV antibody titres (≥ 1:320) were found.
Course in hospital
The patients were in hospital a total of 1856 patient-days, including 532 patient-days in an intensive care unit. The mean length of stay in hospital was 29 days (median 17, range 2 to more than 117 days). Nineteen patients (30%) were admitted to an ICU or a stepdown unit. Sixteen patients (25%) required mechanical ventilation because of a decreased level of consciousness, an inability to handle secretions or respiratory muscle paralysis. The mean length of stay in an ICU was 28 days (median 28, range 2 to more than 83 days [data censored after Dec. 19, 2002). The most common in-hospital complication was pneumonia (in 23% of patients [15/64]). Bacteremia developed in 5 patients (8%), thromboembolic disease in 4 (6%), Clostridium difficile colitis in 3 (5%), myocardial infarction in 5 (8%) and acute renal failure (related to use of contrast agent for CT, blocked continuous bladder irrigation apparatus or medications) in 4 patients (6%).
The outcomes of the 57 patients with neurological manifestations of WNV infection were generally poor. Ten patients died, for a case-fatality rate of 18%. Death occurred a mean of 52 days after admission (range 3–128; data not censored after Dec. 19, 2002). In 8 cases it was attributed directly to one or both of WNV encephalitis or neuromuscular weakness with respiratory failure or aspiration; in the remaining 2 cases death was caused by WNV complicated by pulmonary embolism or sepsis resulting from prolonged hospital stay. Ataxia, weakness and cognitive dysfunction were persistent problems for survivors. At 30 days after the onset of illness, 4 of the 57 patients had died, 37 had persistent neurological deficits and 9 had recovered fully (data missing for 7 patients). The disposition of the 57 patients at discharge is presented in Table 4. Only 13 (23%) (or 28% of the 47 survivors) were discharged home without extra support; many were admitted to rehabilitation facilities or required extra support at home.
Table 4.
Interpretation
The 2002 outbreak of WNV in North America was the largest WNV outbreak ever documented and the largest arboviral outbreak in the Western hemisphere.9 The patients in our series were predominantly active, middle-aged and elderly people living independently in the community. None had been in an institution before their WNV illness, and 72% had been functioning normally and independently (Karnofsky score of 100). Given the central role of mosquitoes in the transmission of WNV, it is intuitive that WNV infection would affect active, relatively healthy individuals, but this idea has not been emphasized in previous reports.7,8,19 The care of the patients reported here was resource intensive, accounting for 1856 acute care hospital days, including 532 patient-days in ICU. This extent of care is an underestimate, because the data for hospital and ICU stays were censored after Dec. 19, 2002. Severe neuromuscular weakness led to prolonged mechanical ventilation in a few cases and weakness and disability in others. As a result, only 28% of those who survived WNV encephalitis or neuromuscular weakness were discharged home without extra support. The 10 deaths were attributed directly to WNV or to complications of prolonged hospital stay that would be expected in older, immobile, obtunded patients.
Pathological study of human autopsy material, including the autopsy reported here, and of primates infected with WNV shows that the virus produces encephalomyelitis of grey matter that involves the cerebral cortex, diencephalon, brainstem (including the cranial nerve nuclei) and the cerebellum.20,21,22,23 Inflammation of the cranial nerve roots has also been reported.23 The majority of patients with encephalitis in our series (65%) had clinical evidence of brainstem, cranial nerve or cerebellar disease, or a combination of these, ranging from dysphagia and ataxia to fatal necrotizing rhomboencephalitis. We believe that the clinical importance of brainstem and cerebellar disease with attendant swallowing disorders and ataxia may have been underemphasized in previous clinical reports.
The hallmark of WNV encephalitis is the combination of encephalopathy with lower motor neuron dysfunction owing to anterior horn cell disease or motor axonal neuropathy, or both. Pathological study of autopsy cases23,24 and experimentally infected monkeys21 suggests that inflammation in the anterior horns of the spinal cord,21,23,24 with motor neuron dropout, is the cause of the characteristic motor neuronopathy. Our finding of nerve root inflammation suggests that this may also contribute to the motor neuronopathy responsible for the weakness. The flaccid paralysis syndrome was described in 19997,8 and again in 2002,24,25,26 when concern was raised that patients were receiving inappropriate therapy aimed at Guillain–Barré syndrome for what was actually anterior horn cell disease or a nonimmune-mediated motor axonal neuropathy.27 In our study, anterior horn cell disease occurred most frequently, but several cases had a combination of axonal and demyelinating neuropathy, and a single case had demyelination alone, which has occasionally been reported previously.28,29,30 Because anterior horn cell disease causes such morbidity and mortality and is not amenable to the treatments used for Guillain–Barré syndrome, efforts must be focused on WNV disease prevention and determining whether agents such as WNV immune globulin31,32 and interferon α-2b33 prevent or attenuate the neurological disease.
The neurological complications of WNV infection most often occur in combination. However, flaccid paralysis, cerebellar or brainstem abnormalities and isolated movement disorders may occur in the absence of a decreased level of consciousness. The variety of neurological pictures we encountered is best represented by the Venn diagram in Fig. 2. Decreased level of consciousness frequently made it difficult or impossible to identify ataxia and neuromuscular weakness. The neurological findings often evolved after discharge from hospital. Therefore, it is important to perform regular follow-up neurological examinations after hospital admission, with particular emphasis on identifying evolving flaccid paralysis and swallowing disorders. CSF examination was useful in identifying meningeal inflammation and excluding other infectious agents. CNS imaging was helpful only in excluding other diagnoses. Serological testing is essential for establishing the diagnosis in individual patients and for timely surveillance of human WNV infection. In our series, the turnaround time for the hemagglutination inhibition assay was often 14 days, and additional time was required for confirmation with the plaque reduction neutralization assay. This delay caused diagnostic uncertainty in individual cases, prompted unnecessary empirical therapy or additional investigation, and delayed recognition of the extent of the WNV outbreak in Ontario. These problems call attention to the need to provide adequate resources for diagnostic laboratory support.
Previous reports have shown that the IgM capture enzyme-linked immunosorbent assay (ELISA) has a sensitivity of 95% and a specificity of 90% with serum specimens taken within 8 days after the onset of illness.34 We need to determine whether this assay will provide an earlier diagnosis than the hemagglutination inhibition assay, which in our series yielded negative results with the first serum sample taken from 38% of the patients. The sensitivity of real-time polymerase chain reaction has been reported to be 55% with CSF samples and 10% with serum samples.34 Our data suggest that the hemagglutination inhibition assay may not detect WNV infection for 2 weeks or more in some immunocompromised patients. Huang and associates35 reported persistent viremia and absent antibody response (determined by means of IgM capture ELISA) in a patient with hematological malignant disease undergoing chemotherapy. Taken together, the results of these and other studies suggest that, in a subset of immunocompromised patients, antibody response is delayed and viremia is prolonged.20,35,36 Therefore, we believe that the IgM capture ELISA should be complemented with nucleic acid amplification tests of CSF and serum samples to achieve the best diagnostic sensitivity early in the course of illness of immunocompromised patients.
The cases we have described emphasize the severe morbidity and mortality associated with WNV infection in the greater Toronto area in 2002. Although our study is limited by its retrospective nature and incomplete case ascertainment, the data provide strong support for an intensive, integrated surveillance and control program for the coming season. Improved and more timely diagnostic testing will be essential for WNV infection surveillance in humans, patient management and blood product screening.
Footnotes
-
Published at www.cmaj.ca on May 6, 2003
This article has been peer reviewed.
Contributors: All authors contributed to the writing of the manuscript and reviewed and approved the final version. In addition, Dr. Pepperell was the principal author, developed the questionnaire, and collected and analyzed data; Drs. Rau, Krajden, Mederski, Burton and Jaigobin helped recognize cases and collected data; Dr. Kern interpreted neurological and neurophysiological findings; Dr. Simor helped recognize cases; Dr. McGeer contributed to the development of the database and the analysis of data; Dr. Mazzulli coordinated the serological testing; Dr. Low contributed to the study concept; Dr. Fearon supervised laboratory testing in Ontario; Drs. Drebot and Artsob supervised laboratory testing in Winnipeg and provided expertise on serologic testing and WNV surveillance in Canada; Dr. Halliday performed the neuropathological examination in the autopsy case and contributed to the interpretation of the results and the writing of the sections on the pathology of WNV infection; Dr. Brunton coordinated the collaboration and collected data.
Competing interests: None declared.
References
In this issue
Article tools






Jump to section
Related Articles
Cited By...
- A Process-Based Model with Temperature, Water, and Lab-derived Data Improves Predictions of Daily Mosquito Density
- Characterization of West Nile Viruses Isolated from Captive American Flamingoes (Phoenicopterus ruber) in Medellin, Colombia
- Elimination of Malaria Risk through Integrated Combination Strategies in a Tropical Military Training Island
- Maculopapular rash and tremor are associated with West Nile fever and neurological syndromes
- Clinical Utility of Commercial Enzyme Immunoassays during the Inaugural Season of West Nile Virus Activity, Alberta, Canada
- The rash of West Nile virus infection
- Cerebrospinal Fluid Neutrophilic Pleocytosis in Hospitalized West Nile virus Patients
- West Nile virus: round five
More in this TOC Section
Similar Articles
Collections

