


In the polymerase chain reaction (PCR), one or more sequences of DNA on a genome or gene transcript are selectively replicated, with up to a billion-fold increase in quantity. The method's power is in allowing the detection or analysis of single or multiple genes in very small samples or in complex mixtures, including the following:
· Identifying parasitic, viral or bacterial DNA from specimens and swabs, including detection of antibiotic resistance.1
· Diagnosing human genetic diseases, including cystic fibrosis, myotonic dystrophy and fragile X syndrome.2,3,4
· Subtyping of malignancies, monitoring response to therapy and detecting recurrence (the sensitivity is sufficient to detect a single tumour cell in one million healthy ones).5
PCR uses specialized enzymes and DNA building blocks to make millions of copies of selected DNA segments in an hour or less. The inventor of the technique, Kerry Mullis, won the Nobel Prize for chemistry in 1993, and PCR made possible the achievement of a draft sequence for the human genome by 2001.
The nitty-gritty
The starting materials for PCR are a DNA “template” molecule, which contains the sequence to be amplified, an enzyme called DNA polymerase (a protein that can create a new DNA molecule), a pair of DNA “primers” and a set of nucleotides (or individual building blocks to be added to the growing DNA molecule by the DNA polymerase). The DNA polymerase is a specially engineered enzyme, which was originally derived from bacteria that live and grow in deep-ocean thermal vents, and hence is resistant to being degraded by temperatures near the boiling point of water. The primers are short (between 10 and 30 nucleotides long) sequences of DNA chosen to bind to opposite ends of the region that is to be amplified.
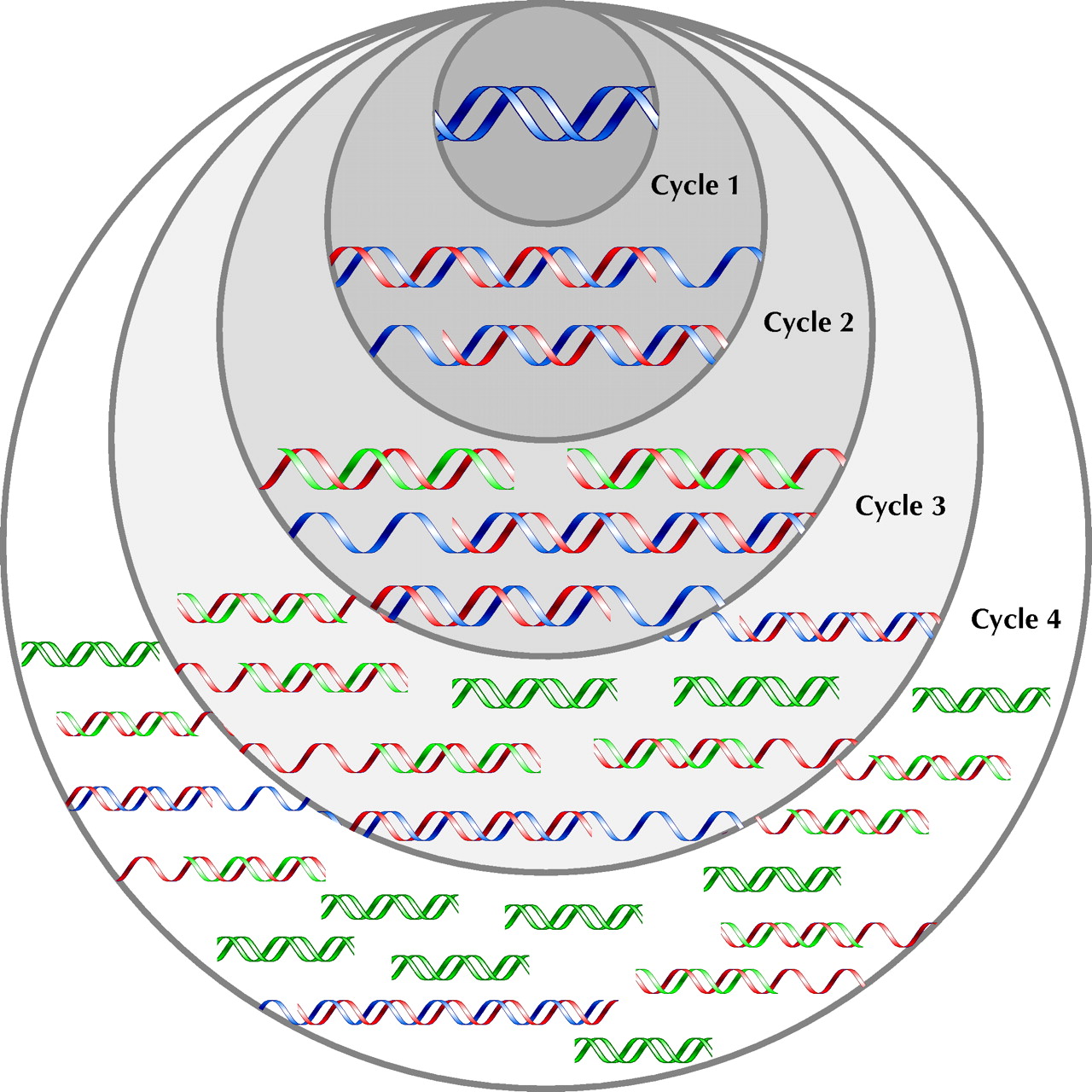
The template, which is ordinarily in the form of a DNA double helix, must be separated into single DNA strands for priming and synthesis. In a living organism, enzymes would separate the strands; in the laboratory, heat is used. The mixture of DNA template, enzymes, primers and nucleotides is briefly heated to 95°C to separate all double helices. It is then cooled to a temperature low enough to allow the primers to bind to their target sequence on the single strands (generally in the mid-40°–60°C range). The temperature is chosen depending upon the sequence and length of the primers, so that only an exact match will allow binding; this gives the method its specificity. Subsequently, the tube is warmed to 72°C for 1–4 minutes to allow the DNA polymerase to synthesize the new DNA strands before being warmed again to 95°C to separate the newly synthesized strands and prepare for another cycle of primer binding and synthesis. Repetition of this cycle results in an exponential increase in the number of copies of the sequence between the 2 primers: 30 repetitions, taking 2 or so hours, will produce a billion such strands (Fig. 1).
{kind=link}
Fig. 1: PCR schematic showing the amplification of the target sequence of DNA (green) from the template DNA (blue). In cycle 1, primers bind to each strand of the template (blue) and DNA polymerase synthesizes 2 new complementary strands (red). There is no signal to stop synthesis, so the daughter strand lengthens until the strands are separated for the next cycle. At the beginning of cycle 2, half the strands are the original template (blue) that synthesize strands of undefined length. However, each strand produced in the first cycle binds the primer directing synthesis in the opposite direction. Synthesis proceeds to a defined end point — the beginning of the strand. The product of this synthesis is a strand of DNA of defined length bounded by the 2 primers (green) and containing the sequence of interest. At the beginning of cycle 3, one-quarter of the strands are the original template (blue). One-half of the strands are single stranded with one defined end (red). Each of these will produce a daughter strand with 2 defined ends (green). One-quarter have 2 defined ends, and each of these will also produce a daughter strand with 2 defined ends (green). With further cycles the original template (blue) cannot increase in number. The strands with one undefined and one defined end (red) can only be synthesized from the original template strands and will increase arithmetically. However the number of strands that have 2 primer-defined ends, and therefore a defined length, will increase exponentially (cycle 4 and beyond).
What then happens to the amplified DNA depends upon how much information is required. If PCR is being used to identify the presence or absence of the target sequence, such as when testing for Chlamydia trichomatis DNA in a urine sample, then it may be detected using the signal of an incorporated fluorescent label, or sought as a band on gel electrophoresis. If the DNA was to be sequenced (read) to determine whether the amplified stretch contained a mutation, such as a specific mutation for cystic fibrosis, it would then be further analyzed for its sequence.
Different applications for PCR exist. Standard PCR allows analysis of genomic DNA (the DNA that is in a cell's nucleus and is a blueprint for all the proteins that the body makes). When a gene is expressed, RNA is generated and this is translated into a protein. “Reverse transcriptase PCR” (RT-PCR) takes the analysis several steps down the pathway toward expression of the gene as protein, as it starts with an RNA template and makes a DNA copy before amplification. Its use is in detection of RNA viruses (HIV or herpes simplex virus) and mutations in genes that affect the editing that occurs in genes at the level of the RNA. “Real-time PCR,” which is currently being developed, promises to allow measurement of the quantity of RNA produced where, for example, a mutation lies in the parts of the gene that control the quantity of RNA transcript rather than its sequence.6
𝛃 See related article page 1021
In this issue
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

