


Abstract
Abstract: Neurofibromatosis type 1 is a common autosomal dominant condition that affects about 1 in 5000 people. We describe a 75-year-old man who, in addition to many classic developmental changes of the disease in his skin, eyes and nervous system, had blindness in his right eye as a complication.
Case: A 75-year-old man with long-standing neurofibromatosis type 1 was admitted because the vision in his right eye had decreased progressively over 3 months. Physical examination showed disseminated cutaneous and subcutaneous neurofibromas of varying size (Figure 1) and café-au-lait spots (Figure 2). The patient had a visual acuity of 6/18 (20/60) in his right eye and Lisch nodules (iris hamartomas) (Figure 3). A neurologic examination showed no abnormalities other than his loss of vision. Axial T1-weighted magnetic resonance imaging of the brain and orbits (Figure 4) showed an isointense mass lateral to the right optic nerve that appeared atrophic and pushed to the left. The mass showed a hyperintense signal on T2-weighted images with contrast enhancement. These findings are compatible with glioma of the optic nerve.

Figure 1: Disseminated cutaneous and subcutaneous neurofibromas of varying size on the torso of a patient with neurofibromatosis type 1.

Figure 2: A café-au-lait spot on the patient's right knee.
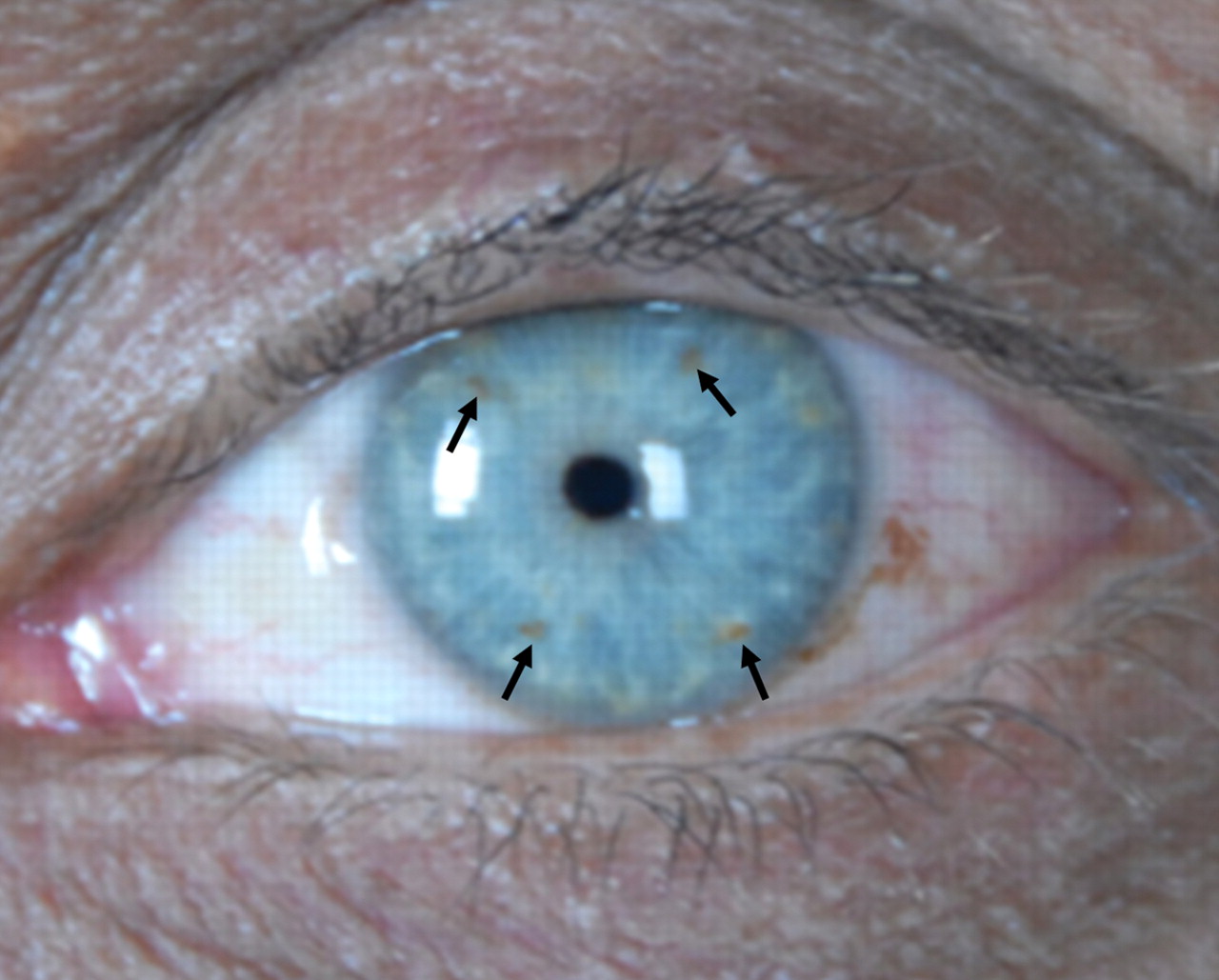
Figure 3: Lisch nodules on the left iris.

Figure 4: T1-weighted axial magnetic resonance imaging of the brain and orbits, showing an isointense mass lateral to the right optic nerve (white arrow) that appears atrophic and pushed to the left (black arrow on inset).
Axial and coronal magnetic resonance imaging (Figure 5) showed a mass in the left parietal lobe with hyperintensity on T2-weighted images and hypointensity on T1-weighted images. After a contrast medium was administered, the lesion showed a thickened, enhanced wall with a central necrotic area. These findings are compatible with astrocytoma.
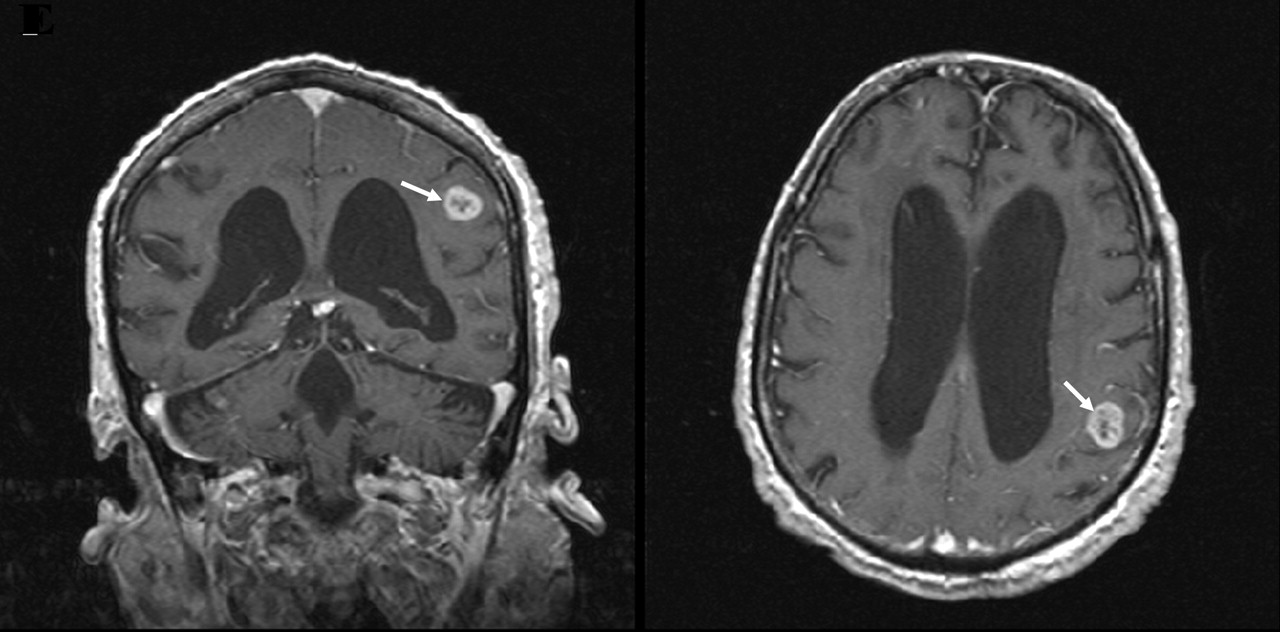
Figure 5: T2-weighted axial (left) and coronal (right) magnetic resonance imaging showing a mass with hyperintensity (arrow) in the left temporal lobe. After administration of a contrast medium, the lesion is visible with a thickened enhanced wall and a central necrotic area.
Because of slight enlargement and increased hardness of the subcutaneous lesions, an excisional biopsy was performed. Histology showed delicate fascicles consisting of cells with oval or spindle-shaped nuclei, scant cytoplasm and round cells with entrapped axons (Figure 6). Only scattered neoplastic Schwann cells were stained during immunostaining for S-100 protein (Figure 7). This pattern is consistent with neurofibroma. The patient chose not to receive further treatment and was discharged.

Figure 6: Biopsy specimen of a subcutaneous neurofibroma showing spindle-shaped and round cells with entrapped axons (hematoxylin and eosin, original magnification ×10).

Figure 7: Only scattered neoplastic Schwann cells (arrow) are stained after immunostaining for S-100 protein. Normally, S-100 protein is present in cells derived from the neural crest, such as Schwann cells. It can be found in melanoma cells, in malignant peripheral nerve sheath tumours and in certain types of sarcomas.
Neurofibromatosis type 1, also known as von Recklinghausen disease,1 is characterized by changes in pigmentation and the growth of tumours along nerves in the skin and other parts of the body. It is caused by a defect in a tumour-suppressing gene on chromosome 17q11.2. Normally the gene produces neurofibromin, a protein that regulates cellular proliferation.2 With the gene mutation, the lack of neurofibromin results in overgrowth of cells from neural crest areas in both the central nervous system (causing Schwann cell tumours on virtually every nerve) and the skin. All people who inherit a copy of the mutated gene are affected. As the pattern of inheritance is autosomal dominant, only 1 copy of the defective gene is needed to cause the condition. However, it is not necessary to have an affected parent. About 30%–50% of patients have a new mutation.
Neurofibromatosis type 2 is a much rarer form of neurofibromatosis caused by mutations in both alleles of a different tumour suppressor gene on chromosome 22q12.1.
About 1 in 3000–5000 individuals are affected by neurofibromatosis type 1, without differences related to ethnic background.3 Pigmented small macules and café-au-lait patches are often present shortly after birth, although neurofibromas are rare in early childhood. In later childhood and adolescence, both neurofibromas and pigmented lesions become common. Clinical manifestations are variable (Table 1).4
Table 1.
A diagnosis of neurofibromatosis type 1 is based on clinical findings. The patient should have 2 or more of the following: 6 or more café-au-lait spots of ≥ 1.5 cm in postpubertal individuals or ≥ 0.5 cm in prepubertal individuals; 2 or more neurofibromas of any type or 1 or more plexiform neurofibroma; and freckling in the underarms and groin.1 The differential diagnosis includes benign café-au-lait pigmentation (present in up to 10% of the general population), multiple lipomas, and sporadic schwannomas, gliomas and meningiomas in the central nervous system.
Most people with mild neurofibromatosis have little disability. People affected by more severe variants have a shortened life expectancy, especially if tumours of the central nervous system or other malignant neoplasms arise during the course of illness.1,3 The condition can have a serious psychological impact because the accumulation of skin nodules can be quite disfiguring.5 Surgical excision and laser treatment of the neurofibromas are possible, but neither treatment is universally effective.6 Transplantation with an allograft of composite tissue on the lower and middle parts of a patient's face was recently reported.7
Gliomas of the optic nerve are found in up to 15% of pediatric patients with neurofibromatosis type 1. Best detected using magnetic resonance imaging, these gliomas are symptomatic in about 50% of patients at diagnosis. A minority will progress to vision loss.8 The high prevalence of gliomas of the optic nerve that are asymptomatic may, however, be biased by referral patterns, Indeed, in patients with neurofibromatosis type 1, the threshold of risk for optic nerve glioma is low.9
Guidelines are available for the diagnosis and management of neurofibromatosis type 1.10,11 Physicians who identify patients with neurofibromatosis type 1 should refer them early to facilities where appropriate evaluation and monitoring of lesions can be carried out. Early detection and monitoring may help to prevent disability and death.
Footnotes
-
Teaching cases are brief case reports that convey clear, practical lessons. Preference is given to common presentations of important rare conditions, and important unusual presentations of common problems. Articles start with a brief summary (100 words) outlining the case and its relevance to a general audience. The case presentation follows (500 words maximum) as well as a discussion of the underlying condition (1000 words maximum). Generally, up to 5 references are permitted and visual elements (e.g., tables of the differential diagnosis, clinical features or diagnostic approach) are encouraged. Written consent from patients for publication of their story is a necessity and should accompany submissions. See information for authors at www.cmaj.ca.
This article has been peer reviewed.
Competing interests: None declared.
Correspondence: Dr. Nicola Mumoli, Department of Internal Medicine, Livorno Hospital, viale Alfieri 36, 57100 Livorno, Italy; fax 39 0586 223251; nimumoli{at}tiscali.it
REFERENCES
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

