


The Case: A 40-year-old woman presented to the emergency department with complaints of hemoptysis and epistaxis that had occurred off and on since childhood, as well as symptoms of dyspnea on exertion, fatigue and oligomennorrhea. She reported a similar history of recurrent epistaxis in her father and both her daughters, but no other significant clinical history.

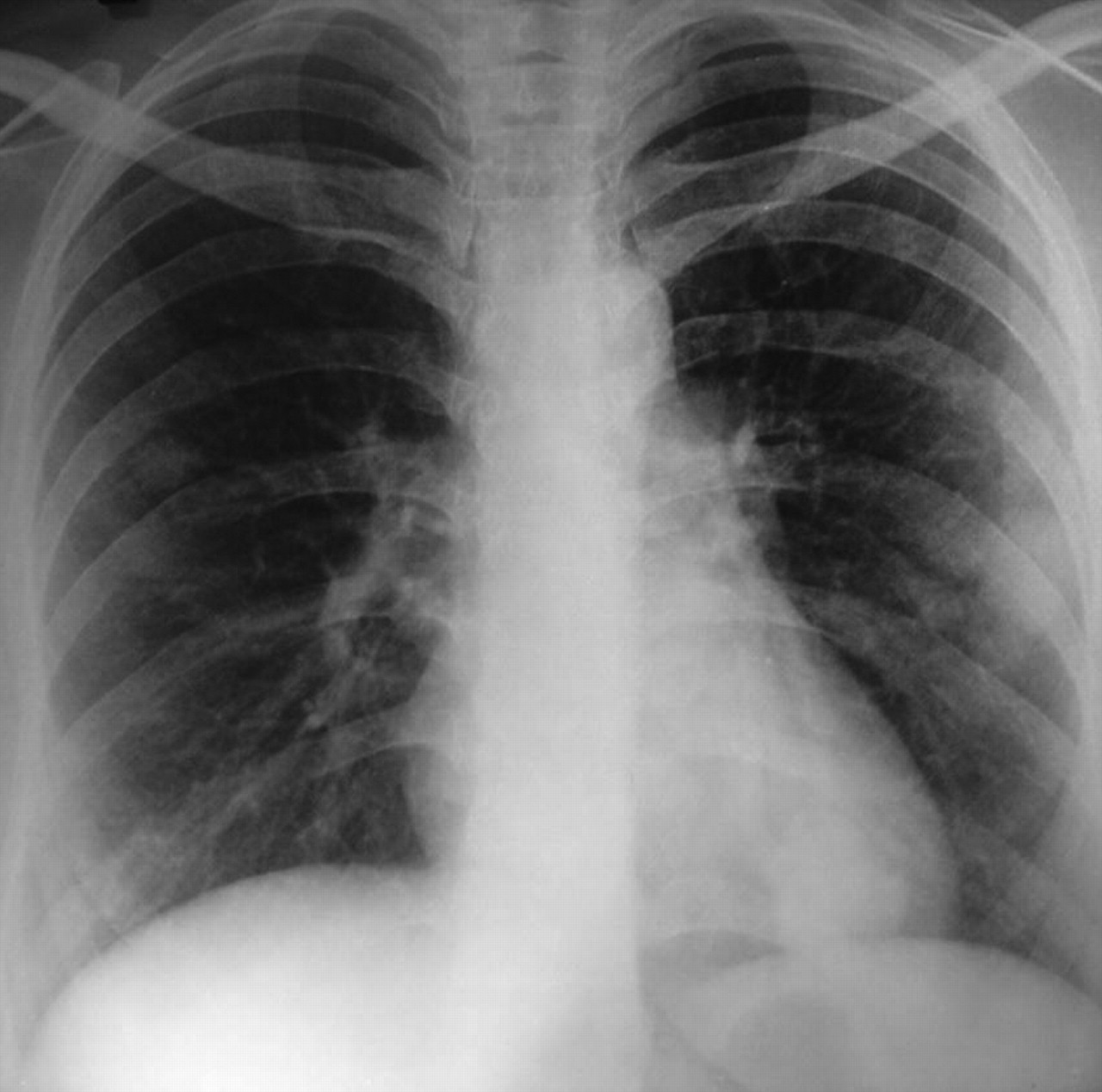
Fig. 1: Chest radiograph, showing multiple nodular shadows in both lung fields in patient with hereditary hemorrhagic telangiectasia (also known as Osler–Weber–Rendu syndrome).
Systemic and gynecologic examinations were noncontributory. Investigations revealed microcytic hypochromic anemia with decreased iron stores. Results of a fecal occult blood test were positive. Radioimaging revealed multiple nodular shadows in both lung fields on a chest radiograph (Fig. 1). CT imaging showed multiple soft-tissue density lesions in the anterior segment of the right upper lobe, and the lateral basal and posterior basal segments of the left lower lobe, with feeding vessels leading into the lesions. Multiple pulmonary arteriovenous malformations (AVMs) were seen on CT pulmonary angiography (Fig. 2A, 2B and 2C). Osler–Rendu–Weber syndrome was diagnosed on the basis of recurrent epistaxis, a strong family history of epistaxis and multiple pulmonary AVMs.

Fig. 2: CT pulmonary angiograms showing arteriovenous malformations (arrows) in (A) apical segment of lower lobe of lung, (B) lateral and posterior segments of lower lobe and (C) lateral and posterior segments of lower lobe.
Osler–Weber–Rendu syndrome, also known as hereditary hemorrhagic telangiectasia (HHT), is an autosomal dominant disorder identified typically by the triad of telangiectasia, recurrent epistaxis and a family history of the disorder. HHT has been subject to underreporting for many years. Recent, careful epidemiologic studies in France, Denmark and Japan, however, have revealed an incidence of 1 in 5–8000. Later descriptions by Osler, Weber and Haynes brought the disorder to the attention of the general medical community.
Currently, 2 loci have been identified associated with HHT: one on chromosome arm 9q33-q34 and a second on chromosome arm 12q. Chromosome arm 9q34 harbours the endoglin gene encoding for a homodimeric integral membrane glycoprotein expressed at high levels on human vascular endothelial cells.
Most commonly, telangiectases involve the mucous membranes, the skin, the conjunctiva, the retina and the gastrointestinal tract. AVMs are found in the lungs, brain and liver. We now estimate that at least 30% of patients with HHT have pulmonary involvement, 30% have hepatic involvement, and 10%–20% have cerebral involvement.
The natural history of HHT has been proposed. It is recognized that the manifestations of HHT are not present generally at birth. They develop with increasing age. Nose bleeds are usually the earliest sign of disease, often occurring in childhood. Pulmonary AVMs become apparent from puberty, and mucocutaneous and gastrointestinal telengiectasias develop progressively with age. Data suggest that, by the age of 16 years, 71% of people with HHT will have some sign of the disease, this proportion increasing to more than 90% by 40 years.1–3
Medical and surgical treatments are aimed at decreasing the amount of hemorrhage and minimizing the sequelae of chronic blood loss. Specific complications, such as brain abscess and high-output cardiac failure, are treated as they arise.
The criteria for the diagnosis of HHT are epistaxis, multiple telangiectases at characteristic sites, visceral lesions (e.g., gastrointestinal telangiectases), pulmonary, hepatic, cerebral or spinal AVMs, and a family history of HHT (based on these criteria) in a first-degree relative. The HHT diagnosis is said to be definite if 3 criteria are present, to be possible or suspected if 2 criteria are present and to be unlikely if fewer than 2 criteria are present.
Our patient presented with a recurrent history of epistaxis, hemoptysis and iron deficiency anemia. On evaluation, she was found to have multiple AVMs and a family history in her father and 2 daughters. A detailed evaluation of her family members is warranted in view of the autosomal dominant inheritance of the disease.
Footnotes
-
Competing interests: None declared.
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

