
Abstract
Cytokines are soluble glycoproteins that are produced by and mediate communication between and within immune and nonimmune cells, organs and organ systems throughout the body. Pro- and anti-inflammatory mediators constitute the inflammatory cytokines, which are modulated by various stimuli, including physical activity, trauma and infection. Physical activity affects local and systemic cytokine production at different levels, often exhibiting striking similarity to the cytokine response to trauma and infection.
The present review examines the cytokine response to short term exercise stress, with an emphasis on the balance between pro- and anti-inflammatory mechanisms and modulation of both innate and specific immune parameters through cytokine regulation. The effects of long term exercise on cytokine responses and the possible impact on various facets of the immune system are also discussed, with reference to both cross-sectional and longitudinal studies of exercise training. Finally, the validity of using exercise as a model for trauma and sepsis is scrutinised in the light of physiological changes, symptomatology and outcome, and limitations of the model are addressed.
Further studies, examining the effect of exercise, trauma and infection on novel cytokines and cytokine systems are needed to elucidate the significance of cytokine regulation by physical activity and, more importantly, to clarify the health implications of short and long term physical activity with respect to overall immune function and resistance to infection.
Similar content being viewed by others

Inflammation ranges from a local to a systemic response to cellular injury. It is characterised by capillary dilatation, production of pathological blood-borne soluble components and an increase in body temperature. Together, these responses initiate the elimination of noxious compounds and damaged tissue. The inflammatory response depends in part on the immune system, which synergises with nonimmune inflammatory mediators such as hepatocytes, neural cells and the endocrine system. Components of the inflammatory response dependent upon and independent of the immune system are induced by stimuli such as physical trauma, exercise and infection. It is not the severity of the injury or insult that governs the symptomatology or outcome of the inflammatory reaction, but rather the mode and extent of response to the original insult.[1]
Cytokines are soluble proteins or glycoproteins that are produced by and mediate communication between and within immune and nonimmune cells, organs and organ systems throughout the body. They act at very low concentrations relative to other soluble molecules such as endocrine and exocrine hormones.[2] The production of cytokines can be up-regulated rapidly in response to inflammatory stimuli, and this response can be transient or prolonged. Circulating concentrations indicate prognosis in various disease and nondisease states.
Modulation of cytokine production by heavy physical activity has many points of similarity with subclinical generalised inflammation. An inappropriate inflammatory response can result in morbidity and even death. The potency of cytokines and the prevalence of cytokine-modulating stressors have prompted the study of stimuli affecting cytokine production. This has led to the suggestion that heavy exercise could be used as a model of trauma and infection.[3,4] The present review briefly summarises the known functions of both pro- and anti-inflammatory cytokines, and it then explores responses to physical activity and training.
1. The Cytokines
1.1 Pro-Inflammatory Cytokines
The pro-inflammatory cytokines and chemokines are up-regulated by physical activity, trauma and infection. Concentrations of the interferons and interleukin (IL)-2 are also modulated by exercise and other inflammatory stimuli, and are thus discussed in the present section. Anti-inflammatory mediators such as IL-4, IL-10, IL-1 receptor antagonist (IL-1ra) and IL-13 buffer the essential but often volatile activities of the pro-inflammatory cytokines, as well as attenuating the immune response.[5] Local and systemic reactions to infection, micro- and macrotrauma depend in part on the magnitude of the cytokine response. The quantitative and qualitative natures of the response dictate outcome; prognosis depends on an appropriate balance between pro- and anti-inflammatory mechanisms.
1.1.1 Interleukin (IL)-1
The IL-1 gene family comprises the glycoprotein receptor agonists IL-1α, IL-1β and IL-1ra. Various immune and nonimmune cells produce both IL-1α and IL-1β. However, IL-1α remains within the cytosol in almost all instances.[6] IL-1α is active in both precursor and mature forms, but IL-1β is only active upon cleavage by the cytosolic cysteine protease IL-1β converting enzyme (ICE), found in monocytes.[7] IL-1β remains cytosolic in nonphagocytic cells, but 40 to 60% of the IL-1β that is produced by mononuclear phagocytes exits the cell via vesicle exocytosis, active transport, leakage or cell death.[6]
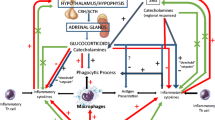
IL-1 elicits physiological effects at femto- to picomolar concentrations, above which it becomes cytotoxic.[8] It up-regulates a wide variety of genes, including those encoding cytokines, thus augmenting its own expression as well as that of IL-2 and IL-6.[5] Enzymes required for the synthesis of leukotrienes, prostaglandins and nitric oxide (NO) are induced by IL-1. Prostaglandin synthesis is induced indirectly, by up-regulation of gene expression of the inducible type 2 cyclo-oxygenase.[6] Cyclo-oxygenase products in turn induce corticotropin (ACTH) release via the hypothalamic-pituitary-adrenal (HPA) axis; this possibly serves as a biological negative feedback, since corticosteroids inhibit pro-inflammatory cytokine gene expression, and adrenalectomised mice have an increased risk of IL-1-induced death.[9]
Responses to IL-1 include hypotension, fever, lethargy, modulation of inflammation and stimulation of cell proliferation. IL-1 induces hypotension by acting on the vasculature, the myocardium, and sodium transport.[10] It inhibits vascular smooth muscle contraction independently of prostaglandin synthesis, facilitating L-arginine-dependent NO production; this leads to increased guanylate cyclase activity.[11] In the myocardium, IL-1 suppresses the rate of spontaneous contractions, and reduces the contractile response to β-adrenergic stimulation.[12] Systemic injection of IL-1 augments the renal excretion of sodium.[6] Within the central nervous system, IL-1 acts as a potent pyrogen, probably at the organum vasculosum of the lamina terminalis; IL-1 receptors are distributed throughout the hypothalamus and arachidonic acid metabolites are released from the periventricular organ endothelium in response to IL-1 injection.[13] Additionally, IL-1 mediates fever both by induction of IL-6 and also independently of the pyrogenic activity of IL-6.[6] IL-1 injection in mice induces disinterest in the environment, sleepiness and anorexia. Presumably, this reaction is analogous to the ‘sickness’ syndrome characteristic of human illness. It may be responsible for many of the symptoms associated with infection and overtraining.
Normal hepatocyte protein production is increased 2- to 3-fold by IL-1, and the synthesis of pathological proteins such as serum amyloid A, C-reactive protein, fibrinogen and leukaemia inhibitory factor (LIF) is increased up to 1000-fold. In this respect, IL-1 synergises with IL-6, but it is also capable of inducing these proteins by itself.[6] IL-1 has inflammatory and degenerative effects on joint surfaces, and systemically injected IL-1 stimulates bone resorption.[14] Aside from inhibiting the formation of collagens and proteoglycans, IL-1 induces synthesis of the metalloproteinases and gelatinases which contribute to the degradation of cartilage. This cytokine also promotes catabolism of lean tissue in synergy with tumour necrosis factor-a (TNFα); muscle protein breakdown during endotoxaemia (which is a potent stimulator of circulatory IL-1 release) can be reduced by treatment with IL-1ra.[15]
IL-1 synergises with the colony stimulating factors (CSFs), IL-3 and IL-6 to regulate the cell cycle and induce lineage-specific and multi-lineage commitment by bone marrow progenitor cells.[16] The effect of IL-1 on immunocompetent cells is that of a cytokine helper.[6] IL-1 amplifies T cell activation by inducing IL-2 and IL-2 receptor (IL-2R) gene expression.[17] Induction of IL-6 (B cell growth factor-2) and proliferation of thymic epithelium are further actions. IL-1 stimulates normal cell and neoplastic cell proliferation.[18] Keratinocytes, mesangial cells, smooth muscle cells and various tumour cells also use IL-1 as a growth factor.[6] IL-1β and TNFα both up-regulate various cell adhesion molecules (CAMs), with implications for the extravasation of cells and metastasis.[6,18]
IL-1ra inhibits the inflammatory effects of IL-1.[8] Intracerebral injection of IL-1ra in mice, for example, completely blocks the pyrogenic effects of a subsequent IL-1 injection.[19]
1.1.2 IL-6
Of several known endogenous pyrogenic cytokines, IL-6 correlates most closely with the degree of fever.[6] This pleiotropic cytokine is produced by various immune cells [such as T and B lymphocytes, natural killer (NK) cells and monocytes] and nonimmune cells (such as smooth muscle cells, chondrocytes, astrocytes and glial cells). It is a glycoprotein of 20 to 30kD molecular mass, depending on its cellular source and method of preparation.[20,21] Various forms of IL-6 have differing biological activities.[21] Cells that express IL-6 receptors include hepatocytes, B cells, T cells, partially committed bone marrow cells, osteocytes and various tumour cell lines.[22] Arguably, the most important effects of IL-6 are upon hepatocytes, B cells and the mononuclear phagocytes responsible for the production of IL-1 and TNF.[23]
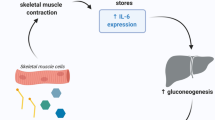
IL-6 is among the most potent mediators of the acute phase response. Other mediators include IL-1, TNF, IL-11, LIF, and oncostatin M.[20,24] The acute phase response is an evolutionarily well conserved cascade of reactions. It is initiated by stressors such as tissue damage, inflammation and heavy exercise. Its objectives are to prevent further damage and activate repair processes. Acute phase proteins synthesised mainly by the liver include serum amyloid A and C-reactive protein.[25] IL-6 enhances ACTH production in the anterior pituitary gland, signalling the adrenal cortex to produce glucocorticoids.[20] The glucocorticoids, in turn, potentiate the effects of IL-6 (and other cytokines) upon hepatocytes. Negative feedback is provided by the inhibitory action of glucocorticoids on macrophages. This further down-regulates the production of IL-6, thus limiting a potentially dangerous positive feedback loop.[24]
The effects of IL-6 on megakaryocytopoiesis and thrombocytopoiesis are well established. In vivo and in vitro studies indicate that IL-6 can act alone or synergise with IL-3 to enhance proliferation of haemopoietic progenitor cells partially or fully committed to the megakaryocytic lineage.[20] Administration of IL-6, either alone or in concert with IL-3, induces a dose-dependent increase in platelet count.[26] IL-6 also functions as a co-stimulatory factor for antigen- or mitogen-induced T cell activation, and is a final maturational requirement for B cells.[27,28] IL-6 further plays key roles in the pathogenesis of various autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis.[29] Finally, it tends to induce bone resorption by selectively activating osteoclasts, a role it shares with other pro-inflammatory cytokines.[29]
Although IL-6 is generally regarded as a pro-inflammatory cytokine, it can play a contrary role. For example, in cancer patients, IL-6 induces the production and release of TNF binding proteins (TNF-BPs) and IL-1ra, thereby buffering the harsh effects of other indisputably pro-inflammatory cytokines.[30] Furthermore, signal transduction through the 130kD glycoprotein receptor subunit gp130 is shared by various cytokines, including the anti-inflammatory IL-11, suggesting a similarity in downstream events.[20,31]
1.1.3 Tumour Necrosis Factor (TNF)
Many of the physiological roles of TNF overlap with those of IL-1 and IL-6, but there are also important points of distinction.[23] TNF is mainly a product of mononuclear phagocytes, but it is also produced by T lymphocytes, Kupffer cells, neural cells and endothelial cells. TNFα and TNFβ have only 30% sequence homology, but exert an overlapping range of effects on the gene expression of growth factors, cytokines, cell-surface proteins and acute phase proteins.[32] Pro-TNFα is cleaved by an unidentified protease to form mature TNFα. Metalloproteinase inhibition can block TNFα secretion in vivo, suggesting that inflammation is closely regulated by the body.[33] Most cells express TNF receptors. Two subunits of these receptors have significant extracellular (TNF-binding) homology, but little similarity intracellularly; this suggests that they have distinct biological activities.[23] Soluble TNF receptors attenuate the potent inflammatory reactions induced by TNF.
As the name implies, TNFα is a powerful tumour necrotising agent, but its therapeutic potential is limited by severe adverse effects.[5] Interferon-γ (IFNγ) synergises with TNFα to mediate tumour cell killing. Like IL-1, TNFα induces the surface expression of various adhesion molecules, [including intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin] thus promoting margination of leucocytes at the site of inflammation.
TNF is also a potent endogenous pyrogen.[34] Presumably, it acts through modification of the physiological temperature set point. However, studies on lipopolysaccharide-induced fever in rats suggests that TNF can also function as an endogenous cryogen or antipyretic.[34,35] Tissue wasting (cachexia) is also a characteristic action of TNFα.[33]
TNFα is rapidly cleared from the circulation by a 2-step process.[36] Firstly, it is inactivated by the binding of TNF-BPs. Thereafter, clearance occurs via the kidneys and to a lesser extent, by metabolism in the liver. TNF-BPs may serve to buffer the human body against the harsh physiological activities of TNFα.[33,37] Release of the p55 and p75 soluble TNF receptors may also reduce sensitivity to the harmful effects of TNF. Furthermore, binding of TNF-BP and soluble TNF receptors to TNF increases the biological stability of TNF, gradually releasing bioavailable TNF into the circulation.
1.1.4 Interferons
The 3 interferons, IFNα, IFNβ and IFNγ, share various properties. IFNα and IFNβ are primarily antiviral agents with some immunomodulatory activity. IFNγ is primarily an immune and inflammatory modulator with 100- to 1000-fold more activity than the other interferons, and it only has secondary antiviral activity.[38] IFNγ is produced by NK cells, CD4+ T helper cells and CD8+ T cytotoxic cells, self-associating in the circulation to form a stable 34kD dimer.[39] It affects both immune and nonimmune cells.
IFNγ is a potent activator of macrophages, and is among the primary cytokines responsible for inducing nonspecific, cell-mediated mechanisms of host defence.[40] It also up-regulates the expression of ICAMs on activated macrophages, thereby contributing to cellular extravasation.[41] It is released by T helper 1 (TH1) cells and is a potent inhibitor of the TH2 humoral immune pathway.[38] NK cell activity is also strongly augmented by IFNγ in a dose-dependent manner, and it increases the sensitivity of tumour cells to NK cells.[42–44]
IFNγ is considered a pro-inflammatory cytokine, in part because it augments the synthesis of other inflammatory cytokines such as TNFα, and in part because it up-regulates expression of the TNF receptors.[45] Another pro-inflammatory role of IFNγ is the induction of at least one form of NO synthase.[46]
IFNγ increases resistance to experimentally induced viral infections, as well as playing a potential role in recovery from infection.[47] Some have explained this phenomenon by implicating IFNγ in an up-regulation of expression of both class I and class II major histocompatibility complex (MHC) proteins.[38] IFNγ also acts directly, by inhibiting viral replication and it serves as an antineoplastic agent.[38] Anti-inflammatory properties may be limited to IFNα and IFNβ.[40]
1.1.5 IL-2
IL-2 is included in this section because of its ability to activate lymphocyte subsets of both the innate and adaptive immune branches, its indirect roles in the inflammatory process, and its relevance to the various inducers of inflammation to be described.[48] It is a glycosylated 133-amino-acid protein produced exclusively by T cells.[49] Three IL-2R subunits exist. The IL-2Rα chain has a low affinity for IL-2, with a dissociation constant (Kd) of 10-8 mol/L, until it joins with the IL-2Rβ chain to form an intermediate affinity complex (Kd 10-9 mol/L). Formation of the high affinity receptor requires complexing of the intermediate affinity receptor with the γ chain; the heterotrimer has a Kd of 10-11 mol/L.[5] Soluble IL-2R (sIL-2R; α and β subunits) attenuate the signal provided by IL-2.[50,51]
IL-2 activates resting T lymphocytes upon mitogen or antigen stimulation. The cytokine acts on T cells in an autocrine or juxtacrine manner.[51,52] Small (naive) T cells strongly express the IL-2Rα chain, but coexpression of α and β receptor subunits occurs in only 1 to 6% of resting T cells.[53] Upon activation with mitogen or antigen, both IL-2Rα and IL-2Rβ become strongly expressed on T lymphocytes. IL-2 induces NK cells to become lymphokine activated killer (LAK) cells. NK cells are a distinct population of CD3e-16+/-56+ lymphocytes that do not rearrange or express the T cell receptor gene; they mediate major histocompatibility complex (MHC)-unrestricted cytotoxicity against a variety of tumour and virus-infected cells.[54] Both CD3e-16-56+ and CD3e-16+56+ cells express IL-2Rβ constitutively at low levels, but only the CD16- subset constitutively expresses the intermediate affinity heterodimeric receptor.[54,55] This subset responds preferentially to lower concentrations of IL-2 with increased cytolytic activity and proliferation. Monocytes and macrophages express IL-2Rβ constitutively at low levels, and they can be induced to express the IL-2Rα receptor subunit upon mitogen stimulation.[56] IL-2 induces proliferation of receptive monocytes, and it also stimulates growth and differentiation of B cells.[57]
IL-15 shares many biological activities with IL-2. Synthesis is primarily by monocytes/macrophages, and unlike IL-2, it is not produced by lymphocytes.[58] The 2 cytokines use common receptor components, but have some distinct actions. For example, like IL-2, IL-15 stimulates B cell expansion, antibody production and proliferation of T and NK cells, but its effects on resting T cells are negligible unless exogenous IL-12 is added.[59]
1.1.6 Chemokines
During the course of inflammation, cells intrinsic to the injured or infected tissue as well as immune cells recruited to the area release chemical attractants (chemokines) that cause leucocytes firstly to adhere to vascular endothelium and subsequently to migrate into the tissue spaces.[5,60] These chemokines play a central role in responses to both disease and heavy exercise.[48,61] They can be divided into 2 families, the CC and the CXC; conserved cysteine residues are separated either by no other residue (CC) or by one other residue (CXC). All of the more than 30 chemokines are small globular proteins of 8 to 11kD molecular mass.[48] The 4 cysteine residues per homodimer are generally conserved between the 2 subfamilies. This allows intrachain disulfide linkage, and contributes to bioactivity and receptor specificity.[60] Like cytokines, the chemokines display synergy, antagonism, redundancy and pleiotropy in chemotaxis. Their spectrum of activities also includes activation and modulation of innate and specific immune cells.
The CXC family is characterised by a short Glu-Leu-Arg motif preceding the first cysteine residue reading N- to C-terminal.[60] IL-8 is representative of the CXC chemokines. It is produced by endothelial cells, fibroblasts, monocytes and macrophages, neutrophils and T lymphocytes, and it is up-regulated by various stimuli including IL-1.[48] Receptors for IL-8 are expressed by neutrophils, monocytes, basophils and NK cells. The primary functions of IL-8 are to attract neutrophils to a site of inflammation or infection and to mediate their activation.[62] IL-8 also attracts T cells and binds to B cell receptors, although these roles are not well understood.
The CC chemokine family includes RANTES (Regulated on Activation, Normal T cell Expressed and Secreted), monocyte inhibitory protein (MIP)-1α, and MIP-1β. The motif is 10 residues in length. Truncation or elongation of this domain leads to a drastic loss in activity, with qualitative changes in remaining activity.[63] The CC chemokines attract nonspecific immune cells such as eosinophils, monocytes and basophils and also T lymphocytes.[48]
1.2 Anti-Inflammatory Cytokines
Inflammation evolved both to protect the organism against infection and injury, and to promote tissue repair. Initially, effector cells are attracted to injured tissue spaces by chemokines produced by intrinsic cells. The next stage involves tissue destruction mediated by the infiltrating cells, with clearance of the damaged tissue. Finally, inflammation subsides, leaving some scar tissue, but allowing a restoration of normal structure and function. Each stage of this process is tightly regulated. The anti-inflammatory cytokines (IL-4, IL-10, IL-13 and perhaps IL-6) attenuate inflammation by restricting inflammatory cytokine production, up-regulating their soluble antagonist binding proteins, and suppressing inflammatory cell activity.[64]
The biological effects of IL-4 are mediated through a dimeric receptor found on most cells of haemopoietic lineage.[65] The cytokine is produced by TH2 lymphocytes, among other cells. It functions to facilitate humoral immunity and inhibits TH1 cells. IL-4 decreases antibody-dependent cell-mediated cytotoxicity and other aspects of macrophage function by attenuating the surface expression of Fc receptors (FcRγI, FcRγII and FcRγIII).[64] IL-4 decreases production of IL-1β, while increasing IL-1ra synthesis. Furthermore, IL-4 induces the cell surface expression and circulatory release of the IL-1 receptor decoy IL-1Rt2; this molecule binds extracellular IL-1 and reduces the synthesis of chemokines such as MIP-1α, but it transduces no signal.[66] IL-4 also reduces the production of NO and reactive oxygen species.[67]
IL-13 is a close structural homologue of IL-4, and it shares some components of the IL-4 receptor upon binding. There is a homology of function for all biological activities on macrophages and neutrophils, but IL-13 has no effect on TH cell proliferation.[64,68,69]
Although IL-6 has some of the properties of a pro-inflammatory mediator (see section 1.1.2), it does not prime vascular endothelium for extravasation of leucocytes, nor does it activate neutrophils or macrophages as do IL-1 and TNF.[64] Several authors have appreciated the anti-inflammatory characteristics of IL-6.[30,64] Its biological activities include stimulation of ACTH release and subsequent elaboration of cortisol (a general anti-inflammatory mediator), with suppression of IL-1 and TNF synthesis by macrophages and neutrophils.[70] IL-6 also attenuates the IL-1 signal by inducing synthesis of IL-1ra and IL-1Rt2, and by promoting the release of these proteins. Overlapping the function of IL-4 and IL-13, IL-6 causes a diminished production of reactive oxygen species and short-lived nitrogenous intermediates, as well as reducing the synthesis of chemokines.[30,64,71]
IL-10, like IL-4 and IL-13, is produced mainly by the TH2 subset of T cells. It is also secreted by macrophages, mast cells and B cells, and is strongly implicated in immunosuppression.[64,71] IL-1 and TNF are potent activators of IL-10 synthesis, thereby inducing a negative feedback mechanism. The anti-inflammatory actions of IL-10 include inhibiting synthesis of the pro-inflammatory mediators IL-1, granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF, IL-8 and IL-12.[64] IL-10 induces macrophages to diminish their NO production, and it inhibits production of IFNγ by T cells and NK cells, thereby retarding development of cell-mediated inflammation.[72] IL-10 also promotes proliferation of B cells and mast cells, and it acts on monocytes to induce production of IL-1ra and the soluble TNF p55 receptor.[73,74]
2. Cytokine Response to Physical Activity
If physical activity is of sufficient vigour to induce an inflammatory response, there is a release, firstly of a sequence of pro-inflammatory cytokines (TNFα, IL-β and IL-6), and then of regulatory, anti-inflammatory cytokines (e.g., IL-4, IL-10 and IL-1ra).[75]
2.1 Effects of Short Term Physical Activity on the Pro-Inflammatory Cytokines
Studies examining cytokine production in the context of prolonged bouts of deliberate endurance exercise are most common, but additional insights have been gained from other forms of short term physical activity such as near maximal/short duration exercise, anaerobic exercise and concentric and eccentric activity. The response differs appreciably with the mode of exercise,[76–79] the availability of carbohydrate[79] and core temperature.[80]
Until recently, the source of the activity-induced increase in serum cytokine levels remained speculative. Bruunsgaard et al.[78] found messenger RNA (mRNA) for IL-6 in exercising muscle, but not in the blood. Moldoveanu et al.[81] also found no change of mRNA levels for IL-1β, IL-6 and TNFα in circulating cells in response to 3 hours of moderate endurance exercise. However, flow cytometry has now demonstrated that an increased percentage of circulating monocytes express pro-inflammatory cytokines intracellularly during and immediately following vigorous exercise,[82] suggesting that these cells are at least one source of the cytokines.
Table I summarises the effects of exercise on some of the pro-inflammatory cytokines.

Effects of short term exercise on pro-inflammatory cytokines.

Table I. Contd.
2.1.1 IL-1
Plasma concentrations of IL-1 are particularly difficult to measure, as basal circulatory levels remain at the sensitivity threshold of available technology. In consequence, results are inconsistent.[2]
Northoff et al.[112] found modest increases of IL-1 following exercise, with an associated increase in urinary excretion. Moldoveanu et al.[81] had individuals exercise for 3 hours at 60 to 65% of maximal oxygen intake (V̇O2max); they noted a significant increase of IL-1 at 60 minutes, with a further increase at 180 minutes; values remained above initial levels for 24 hours following exercise. Ostrowski et al.[113] also saw a 2.1-fold increase of IL-1β in the first hour after running a marathon race. On the other hand, Suzuki et al.[93] found no increase of IL-1β following a marathon run. Bruunsgaard et al.[78] used an eccentric exercise protocol consisting of 30 minutes of maximal and supramaximal-equivalent cycle ergometry in an attempt to investigate the effects of such stimuli on circulatory IL-1β. However, plasma IL-1β levels as determined by enzyme-linked immunosorbent assay (ELISA) remained below the detection threshold for all sampling time-points.
Ullum et al.[88] studied IL-1β gene expression in 17 healthy, moderately trained males in response to 60 minutes of cycle ergometry at 75% of each individual’s V̇O2max. Using nuclear run-off analysis, they noted no change in pre-mRNA isolated from peripheral blood mononuclear cells (PBMC) before, during or after exercise. In accordance with this observation, Natelson et al.[90] observed no change in PBMC RNA levels of either IL-1α or IL-1β after a staged walk exercise bout that progressed in speed and inclination for 46 minutes, to the point of voluntary exhaustion.
Cannon et al.[86] investigated the intramuscular IL-1β level in 5 healthy males. Eccentric exercise was achieved by running downhill on a 16% treadmill slope for 45 minutes at a speed demanding 75% of the individuals’ concentric V̇O2max. Muscle biopsies were stained immunohistochemically with rabbit antisera raised against human IL-1β, and the investigators also used a T cell proliferation (inducible by IL-1β) bioassay. Intramuscular IL-1β concentrations were increased at 45 minutes and 5 days after exercise.
Although observations of IL-1 protein production in response to exercise appear inconsistent with unchanged IL-1 gene expression, extracellular cytokine release is not always secondary to the immediate gene expression of a given cytokine. Mast cells, for example, store TNFα until they degranulate.[114] Thus, sources of IL-1 such as the mononuclear phagocytes may be storing IL-1 intracellularly before its extracellular release.
Some investigators have looked at the IL-1 response to mitogenic stimulation following exercise. Lewicki et al.[92] observed that 48 hours of stimulation by lipopolysaccharide (40 mg/ml) increased the concentration of nonexercised PBMC supernatant IL-1 to 27.4 units/ml and that of exercised PBMCs to 48.7 units/ml. Moyna et al.[115] increased the cycle ergometer loading every 6 minutes in highly trained cyclists (32 males and 32 females). Consecutive stages of exercise amounted to 55%, 70% and 85% of the individual’s V̇O2max. Lipopolysaccharide-induced in vitro IL-1β production (ELISA methodology) increased throughout the 18 minutes of exercise, but had returned to baseline 2 hours after exercise. Haahr et al.[83] measured unstimulated and lipopolysaccharide-stimulated IL-1α and IL-1β production, using the ELISA method and a thymocyte proliferation bioassay. Both techniques showed that 2 hours after 60 minutes of 75% V̇O2max cycle ergometry, lipopolysaccharide-induced IL-1 production was augmented. In apparent conflict with these results, Weinstock et al.[94] demonstrated that 68 minutes of competition-intensity triathlon exercise decreased the ability of stimulated whole blood cultures to produce IL-1β. However, this study did not account for the possible effects of competitive stress on cytokine production.
Others have examined the effects of exercise on unstimulated IL-1β production. Sprenger et al.[95] examined the excretion of IL-1β in 22 distance runners who covered a distance of 20km in roughly 110 minutes. Urine concentrations of IL-1β increased about 1.8-fold relative to before exercise, and values remained high for at least 24 hours following exercise. Bury et al.[96] had 7 healthy males exercise for 4 hours at 45% V̇O2max, 3 hours at 60% V̇O2max, and 2 hours at 75% V̇O2max, each bout of exercise being separated by enough time to preclude a cumulative training effect. With each of these protocols, IL-1 levels were significantly elevated, both immediately following exercise and 60 minutes after its termination. The magnitude of the increase depended on the intensity rather than the duration of exercise. Many other observers have found that IL-1β production is unaffected by exercise unless the blood is stimulated.[110,116,117] It appears that although exercise alone may induce a limited output of IL-1, subsequent mitogenic stimulation allows investigators to amplify the effects of exercise, with implications not only for detection of this cytokine, but also for immune activation by exercise. IL-1 release depends on the type of exercise performed, its intensity and, to a lesser degree, the duration of exercise. IL-1 is among the first cytokines to be released in response to exercise stress.
2.1.2 IL-6
Exercise generally causes a strong but transient induction of IL-6. The magnitude of the response depends on the intensity of effort, and values typically normalise within 6 hours or less.[75–77,79,81,94,95,101,102,110,112,116,118] Suzuki et al.[104] have drawn attention to a close correlation between exercise-induced neutrophilia and increases of IL-6, and they speculate that a stress-induced release of bioactive substances may mobilise and prime neutrophils, causing local tissue damage and an inflammatory response.
Many investigators have examined the unstimulated and stimulated release of IL-6 in response to various forms of exercise. Rivier et al.[100] investigated IL-6 levels in the circulation as well as in the supernatant of lipopolysaccharide-stimulated monocytes in response to a cycle ergometer test of peak aerobic power. The participants were a heterogeneous group of masters and younger athletes. Stimulated IL-6 release was moderately increased immediately after exercise, but had reverted to resting values after 20 minutes of recovery. Moldoveanu et al.[81] exercised young volunteers for 3 hours at 60 to 65% of V̇O2max; this induced an 18-fold increase of IL-6, and values remained above initial resting levels for at least 2 hours. Ullum et al.[88] had individuals exercise at 75% of their individual V̇O2max for 60 minutes. They found that plasma IL-6 levels rose significantly only during exercise; 2 hours after exercise, IL-6 concentrations had dropped back to baseline values.
Gannon et al.[101,102] reported major increases of IL-6, 10 to 25 and 150 minutes following a 6-hour cycle race. The urinary IL-6 response to a 60-minute triathlon was investigated by Northoff and Berg.[103] They found elevated levels of IL-6 immediately after the run, but did not examine IL-6 concentrations at subsequent time-points. Northoff and Berg[103] also quantified cytokine release in the plasma of marathon runners. In this population, they found a significant increase shortly after the run, but not 24 hours later. Ostrowski et al.[85] had 10 moderately fit individuals exercise on a treadmill for 2.5 hours at 75% of their individual V̇O2max, and they also analysed plasma from athletes competing in the Copenhagen marathon. In accordance with the other studies, plasma IL-6 peaked immediately after exercise in both cases. Suzuki et al.[93] also observed 100-fold increases in IL-6 immediately following a marathon race. Northoff et al.[119] used a bioassay to semi-quantify plasma IL-6 concentrations in long-distance runners. Compared with pre-run values (37.6 units/ml), circulating IL-6 levels were significantly higher immediately after a prolonged run (79.8 units/ml), but not 24 hours later (41.4 units/ml). Unfortunately, although the proliferative bioassay used in this early study was sensitive to IL-6, it was not proven to be insensitive to IL-7. Nehlsen-Cannarella et al.[98] showed that plasma IL-6 levels in 30 experienced marathon runners peaked immediately after a 2.5 hours run, whether individuals were carbohydrate-supplemented or not; values had returned close to baseline 1.5 to 3 hours after exercise.
Subsequent observations by the same group showed that IL-6 levels were lower for cycling than for a similar intensity of running, and were further reduced by carbohydrate supplements.[79] Bruunsgaard et al.[78] compared values for normal and eccentric cycling, finding that the latter induced a larger secretion of IL-6. Brenner et al.[76,77] also found a significant increase of plasma IL-6 with 2 hours of endurance exercise at 60% of peak aerobic power, but there was no change with 5 minutes of exercise at 90% of peak aerobic power, or with an exhausting circuit-training routine.
Papanicolaou et al.[99] reported a biphasic IL-6 response after 25 minutes of intermittent treadmill running at a high relative intensity; 15 moderately trained males warmed up with 5 minutes of light running (50% V̇O2max) and completed 10 brief bouts of inclined treadmill running (each lasting 30 seconds at 90% V̇O2max), followed by a 10-minute cool-down of slow jogging. Plasma IL-6 peaked twice: once at the end of the high intensity exercise, and once 20 minutes after exercise.
Heterogeneous studies thus point to the conclusion that circulatory concentrations of IL-6 are elevated rapidly but transiently in response to various types, durations and intensities of exercise. Tissue damage has been implicated in the induction of blood-borne and tissue IL-6.[78,89] Natelson et al.[90] used their aforementioned graded exercise protocol to induce temporary exhaustion; IL-6 RNA expression was unchanged by this exercise. Likewise, neither Ullum et al.[88] nor Moldoveanu et al.[81] found any up-regulation of IL-6 gene expression in mononuclear cells response to exercise. However, as discussed for IL-1β, flow cytometry shows increased intracellular levels of IL-6,[82] and circulatory release does not necessarily depend on immediate induction of cytokine gene expression.[114,120,121] For this reason, there is a need for further studies relating the kinetics of cytokine appearance to the intracellular concentrations found in various tissue compartments.
2.1.3 TNF
Suzuki et al.[93] were unable to detect TNFα in the plasma following a marathon run, perhaps because they collected samples immediately following the event. Most other investigators have found that prolonged endurance exercise induces TNF release into the plasma. Rokitzki et al.[105] detected increased TNFα levels in the plasma of 14 endurance-trained athletes immediately following a marathon run. Dufaux and Order[106] followed the kinetics of plasma TNFα in 8 male sports students after a 2.5-hour run. Peak levels of this cytokine were reached 60 minutes after the end of the race (the only statistically significantly difference), but concentrations had fallen below initial control values 120 minutes after the end of the race. In accordance with this observation, Ostrowski et al.[85] showed a 2.5-fold increase in plasma TNFα levels immediately after 2.5 hours of treadmill running at moderate-to-high intensity.
Brenner et al.[76,77] found an increase of TNFα in response to 2 hours of cycle ergometer exercise (60% of V̇O2max), with peak levels reached 72 hours after exercise. Likewise, Moldoveanu et al.[81] observed a 90% increase of plasma TNFα following 3 hours of endurance exercise at 60 to 65% of V̇O2max; this abated gradually over the following 24 hours. However, no significant changes were induced by 5 minutes at 90% of V̇O2max, or by an exhausting circuit training routine. Sprenger and associates[95] found that the urinary excretion of TNFα was elevated immediately after a 20km run, but the effect was transient; levels became normal 1 or more days after the run. Espersen et al.[107] similarly found that a 5km race was sufficient to elevate plasma TNFα levels immediately after effort.
Rivier et al.[100] investigated TNFα production by blood monocytes in terms of translated and secreted protein. Combined data from masters athletes and younger individuals indicated a nonsignificant trend for monocyte TNF release to increase immediately after exercise, but in their participants there was a decrease below baseline during the post-exercise recovery period. Further evidence that exercise may attenuate monocyte TNF production comes from the work of Bagby et al.[109] They investigated the effect of exhaustive running-wheel exercise on the TNF response of rats to lipopolysaccharide (1 mg/kg body mass). Exercise severely depressed the circulatory TNFα response for up to 3 hours after exercise. Moreover, heart rate and blood pressure nadirs correlated with TNFα concentrations, recapitulating the effects of stress-activated TNFα on the myocardium and vasculature.[122] Baum et al.[108] noted that 90 minutes of exhaustive exercise decreased the release of TNFα in lipopolysaccharide-stimulated whole blood. Exercise, therefore, appears to have different effects on the production of IL-1 and TNF by immune cells; it tends to magnify the IL-1 response to lipopolysaccharide, but it can reduce that to TNF. This is surprising, given the close association of these 2 cytokines in pathophysiological contexts such as general inflammation. Divergent results in studies focusing on TNF may be explicable by inconsistencies in experimental design. In vivo or in vitro administration of glucocorticoids suppresses the lipopolysaccharide-induced release of TNFα by blood monocytes.[123] It is though that elevated glucocorticoid levels suppress the effect of exercise on circulatory TNF, and it may be instructive to test this hypothesis in adrenalectomised animals.[123] In humans, investigation of the dependency of TNF on cortisol levels may be enlightening, as would be protocols designed to account for or eliminate the glucocorticoid stress response.
Ullum et al.[88] found no change in the quantities of TNFα pre-mRNA isolated from peripheral blood monocytes in response to physical activity. Likewise, Moldoveanu et al.[81] saw no increase of TNFα mRNA in mononuclear cells, and Natelson et al.[90] demonstrated that 45 minutes of progressive treadmill exercise to exhaustion attenuated TNFα gene expression by PBMC isolated immediately after exercise. The reverse transcription polymerase chain reaction (RT-PCR) was used to show that of the pro-inflammatory cytokines, only TNFα gene expression was affected by exercise.
In summary, human TNF modulation by exercise seems to be influenced by intensity and particularly by duration of the exercise stimulus. Training may also have an effect on the TNFα response (see section 3). Exercise has either no effect or a slight inductive effect on circulating TNFα, although the effects on monocyte and PBMC TNFα production indicate that circulatory TNF up-regulation may not be the result of PBMC TNF release.
2.1.4 Interferon-γ
Studies of the effect of exercise on IFNγ levels to date have been largely equivocal (table II).[2] Haahr et al.[83] had individuals exercise at 75% of maximal aerobic power for 60 minutes; they could not detect any change in circulating IFNγ levels. Likewise, Gannon et al.[101,102] were unable to detect IFNγ in the plasma following 6 hours of competitive cycling, and Suzuki et al.[93] found no change of IFNγ following completion of a marathon run.

Effects of exercise on interleukin (IL)-2, IL-2 receptor (IL-2R) and interferon-γ (IFNγ).
The graded treadmill exercise protocol employed by Natelson et al.[90] did not produce any significant change in IFNγ gene expression in peripheral blood lymphocytes. At the level of IFNγ protein release, the 90-minute exhaustive exercise protocol adopted by Baum and associates[108] transiently decreased the ability of lipopolysaccharide-induced whole blood cultures to produce IFNγ. More moderate physical activity (cycle ergometry at 70% of individual anaerobic threshold for 30 minutes) induced a quantitatively similar attenuation in the ability of phytohaemagglutinin (PHA)- and staphylococcal enterotoxin B-stimulated whole blood cultures to produce the IFNγ protein.[108] However, Surkina et al.[132] determined that in highly trained male rowers, 7 minutes of race simulation decreased the ability of isolated lymphocytes to produce IFNγ.
The data thus far indicate that exercise has either little effect or attenuates IFNγ synthesis. A decrease in IFNγ response to mitogenic stimulation, and thus lower plasma concentrations of this cytokine has been suggested as contributing to an ‘open window’ for infections following exercise.[132,133] However, at least one group of investigators[130] has shown that plasma isolated from exercised individuals has an increased antiviral activity after exercise. Viti et al.[130] used a bioassay sensitive to IFNα to investigate the effects of 60 minutes of 70% V̇O2max cycle ergometry on plasma release of this cytokine. IFNα levels increased only 2-fold after exercise and remained at this level for another hour before falling back to baseline. More research is needed to clarify discrepant results, to understand further the mechanism predisposing competitive athletes and overtrained individuals to infection, and to determine the relationship of any changes in susceptibility to interferon levels.
2.1.5 IL-2
Most studies show that exercise impairs the ability of mitogenically stimulated lymphocytes to produce IL-2. Data on unstimulated cells are more divergent (table II). Rhind et al.[124] reported that 60 minutes of exercise at 60% V̇O2max decreased culture supernatant IL-2 production by PHA-stimulated PBMC; no IL-2 production was detected in unstimulated cells. The [3H]thymidine incorporation of PHA-stimulated cells, indicative of DNA synthesis, was also reduced by exercise. Administration of recombinant IL-2 (rIL-2) enhanced PHA-stimulated cell growth, but did not reverse the exercise-induced suppression of mitogen responsiveness. However, exercise made the cells more responsive to rIL-2 administration, an observation explicable by changes in soluble and cell surface IL-2R. In agreement with these data, Lewicki et al.[92] showed that when 11 trained men were exercised incrementally to exhaustion (over a mean time of 20 minutes), the PHA-stimulated IL-2 release of PBMC was diminished by 27% immediately after exercise and suppression was even greater 2 hours after exercise. Espersen et al.[107] were able to measure plasma IL-2 levels. They found no change immediately after exercise, but plasma IL-2 levels had increased significantly over baseline 24 hours after exercise. Animal studies on splenocyte IL-2 production generally have used concanavalin-A (Con-A) stimulation, but results have failed to agree with each other.
Studies investigating cell surface IL-2R have looked at one or more of the 3 receptor subunits, whereas measurements of sIL-2R have usually been made on IL-2Rα (table II). Rhind et al.[124] demonstrated that serum IL-2R levels were elevated significantly immediately after exercise, and peaked 60 minutes after cessation of physical activity. The IL-2Rα (CD25) and IL-2Rβ (CD122) subunits of IL-2R were transiently up-regulated on the lymphocyte cell surface during exercise. Possibly, this could account for the increased proliferation in response to rIL-2 in lymphocytes taken from individuals who had exercised. The CD25+ (IL-2Rα+) and CD122+ (IL-2Rβ+) phenotypes diminished to normal levels within 30 minutes of recovery.[124] Dufaux and Order[106] showed a more prolonged increase in sIL-2R levels in response to a 2.5-hour run. Levels rose during the first 24 hours of recovery, although increments only became statistically significant at 24 and 48 hours of sampling. Lewicki et al.[92] showed that after incremental exercise to exhaustion, PHA stimulation of PBMC resulted in a 23% decreased expression of CD25, whereas the CD25 expression of unstimulated cells increased 3-fold in response to exercise. Sprenger et al.[95] noted that under cold conditions, a 20km run lasting 95 to 120 minutes doubled urinary excretion of sIL-2R.
Since IL-2 is an important mediator of T cell activation, such exercise-induced changes may mechanistically affect adaptive immune cell-mediated cytotoxicity. The increased proportions of PBMCs that express the CD25 and CD122 antigens suggest that short term exercise enhances cellular immunity. On the other hand, reproducible decreases in the ability of PBMC to respond to polyclonal stimulation by synthesising IL-2 implies that exercise suppresses some aspects of cell-mediated immunity. Further work is needed to determine more holistically the effects of exercise on cell-mediated immune function through modulation of IL-2 and its receptor subunits.
2.1.6 IL-8
Although IL-8 is the only chemokine that will be discussed in the context of exercise, this chemokine is not necessarily representative of all chemokines. Other cytokines have different functions and accordingly different modulatory mechanisms. Because of the chemotactic properties of IL-8, its modulation by exercise may contribute to the mobilisation of immune cells during physical activity.
Nielsen and co-workers[134] used a 2-hour rowing ergometer protocol to determine whether a decrease in pulmonary diffusion capacity after exercise was a precursor of an adult-onset respiratory distress syndrome (ARDS)-like injury. That exercise may induce an ARDS-like injury is supported by observations from various authors who have found an increased fluid volume in the lungs after prolonged exercise. Plasma IL-8 measurements remained below the detection threshold before, during and after exercise, suggesting that some other factor had altered pulmonary diffusion capacity. Hopkins et al.[135] were able to measure IL-8 reliably. No changes in circulating IL-8 concentrations were seen 60 minutes after a high-intensity 7-minute bicycle race. The one positive finding to date is from Suzuki et al.,[93] who observed a significant increase in IL-8 after individuals had participated in a marathon event.
Since exercise does not generally increase circulatory IL-8, it is possible that IL-8 production may be a strictly local phenomenon, with only a limited spill-over into the systemic circulation relative to the biological half-life of this chemokine. It remains important to investigate the impact of other chemokines on cellular migration, in the hopes of describing mechanistically the pronounced leucocytosis and lymphopenia induced during and following exercise, respectively.
2.2 Effects of Physical Activity on the Anti-Inflammatory Cytokines
2.2.1 IL-1 Receptor Antagonist
Table III summarises the effects of exercise on 3 anti-inflammatory cytokines. Exercise induces a 2- to 214-fold increase in circulatory IL-1rα levels.[75,93,98,110] In elite runners, a 6-hour endurance race induced a 5-fold increase in plasma IL-1rα immediately after the event.[110] Ostrowski et al.[85] also found that 2.5 hours at 75% V̇O2max increased circulatory IL-1rα levels 78-fold 1 to 2 hours after exercise. The largest increase, 214-fold, was found immediately following participation in a marathon race.[93]

Effects of exercise on anti-inflammatory cytokines.
In agreement with these observations, Nehlsen-Cannarella and associates[98] found that triathletes who were not carbohydrate-supplemented increased their plasma IL-1rα roughly 2-fold 90 minutes after a 2.5-hour endurance run. Levels were decreased by provision of carbohydrates, but did not differ between cycle ergometer and running exercise.[79]
2.2.2 IL-4
Moyna and colleagues[115] found that 18 minutes of cycle ergometry increasing from low to moderate intensity did not affect plasma IL-4 levels. Likewise, Suzuki et al.[93] found no increase of plasma IL-4 immediately following a marathon run. Unfortunately, no one has yet examined the effect of exercise on IL-13 production.
Natelson et al.[90] found that short, near-maximal treadmill exercise did not affect the gene expression of IL-4 by PBMC, as measured by RT-PCR. Others have used RT-PCR to evaluate the effects of 30 minutes of cycle ergometry at 70% of maximal aerobic power; qualitative results suggest that IL-4 mRNA levels are unchanged in response to this stimulus.[137]
2.2.3 IL-10
IL-10 has been implicated by many investigators as effecting the immunosuppression associated with various forms of trauma, including exercise.[3,138] Brenner et al.[76] had untrained males exercise using 3 different protocols. Neither 2 hours of cycle ergometry at 65% of peak aerobic power nor 45 minutes of intermittent resistance exercise had any impact on plasma IL-10 concentration. However, 5 minutes of intense exercise (90% of V̇O2max) decreased plasma IL-10 concentrations 3, 24 and 72 hours after exercise, although the change was small and its biological significance is likely to have been negligible.[138] Gannon et al.[101,102] found no measurable increase in IL-10 following 6 hours of competitive cycling, but Suzuki et al.[93] found a 3.4-fold increase in plasma IL-10 immediately following a marathon run, and Anwar et al.[91] found that serum IL-10 was elevated significantly 48 and 72 hours after strenuous eccentric exercise.
Changes in de novo gene expression of IL-10 may not be responsible for any changes in circulating IL-10, at least after short, near-maximal exercise. Natelson et al.[90] found that peripheral blood leucocyte RNA levels of IL-10 were unchanged after a maximal treadmill stress test. Although IL-10 is greatly affected by other forms of trauma, it remains to be determined how far exercise-induced changes in concentrations of this cytokine are physiologically relevant. The appearance of IL-10 after eccentric exercise may indicate that IL-10 release is secondary to tissue damage.
3. Effects of Training on the Cytokine Response
The ability of exercise to modulate immune function through cytokine production has prompted some investigators to explore the effects of training on immune status.[1,139–143] However, few studies have looked at cytokine responses. This reflects logistical problems associated with well controlled longitudinal studies.
Early cross-sectional studies relating immune status to level of training or fitness revealed some important trends. IL-2 and its receptor subunits have been afforded unrepresentative attention. The observation that aging has a suppressant effect on cellular immune function has prompted training studies in the elderly. These have focused in part on the cytokine response, in the hope of developing strategies for counteracting immunosenescence.[143] Cross-sectional and longitudinal studies have also investigated the effects of training on the cytokines that mediate inflammation. As cytokines modulate cell-mediated immunity directly, determining the immune and nonimmune cellular response to training has allowed an indirect insight into the cytokine response and adaptation to long term physical activity. The effects of training on humoral immunity are beyond the scope of this review, but the effect of training on cytokines supportive of humoral function has received little study to date.
Baum et al.[139] had individuals train by 3 to 5 hours of field running per week for 12 weeks. This induced an increased synthesis of IL-1β and IL-6, but no changes in IL-2, sIL-2 or IFN-γ.
Baj et al.[144] followed a cohort of male cyclists for 6 months of competitive endurance training. IL-2 concentrations in the supernatant of PHA-stimulated PBMC isolated under resting conditions were attenuated by approximately 80% as the competitive season advanced.
Rhind and co-workers[124] examined the effects of a 12-week programme of moderate training. Individuals performed 30 minutes of cycle ergometry at 65% to 70% of maximal aerobic power, 4 to 5 times per week. Although 60 minutes of aerobic exercise decreased PHA-stimulated IL-2 production in PBMC culture supernatant in both the untrained and trained state, 12 weeks of training significantly attenuated the suppression of IL-2 production. [3H]Thymidine incorporation indicated that cellular proliferation was greater in the trained group during and after exercise, possibly indicating greater IL-2 production and/or receptor expression. Others have found no influence of training on IL-2 production; Weicher and Werle[145] reported that 4 months of training did not change circulatory levels of IL-2 at rest or after short term exercise.
As in short term exercise studies, the effects of training on the receptor end of the IL-2 signal transduction pathway has received more attention than the IL-2 ligand itself. Studies have focused either on circulatory CD25 or cell surface expression of both CD25 and CD122. Gray et al.[129] found that 1 minute of high-intensity cycling in highly trained cyclists significantly decreased CD25+ cell counts 6 hours after exercise. However, this effect was not observed in sedentary controls. Resting levels of CD25+ PBMC were notably lower in 7 trained athletes after in vitro stimulation with pokeweed mitogen.[146]
Animal studies have yielded equally variable results.[2] Rhind et al.[124,147] used a 12-week training protocol to gauge training effects on human CD25 and CD122 surface expression. 30% of circulating lymphocytes expressed the low affinity receptor CD25 under resting conditions in both untrained and trained states. However, trained individuals had an increased CD25+ cell count 60 minutes after exercise, whereas the untrained had a depressed CD25+ count at this same time. The results for the IL-2Rβ subunit were more dramatic: before training, 10% of unstimulated lymphocytes expressed CD122, whereas after training the corresponding concentrations of CD122+ cells had increased by 36%. This study showed that resting levels of soluble CD25 increased by 33% following training and that training altered the kinetics of sIL-2R release during 60 minutes of cycle ergometry.
4. Aging and the Cytokine Response
Aging induces a profound decrease in cellular immune function. This is mediated in part by changes in cytokine response to various stimuli. Although peripheral blood concentrations of some pro-inflammatory cytokines increase with age, others generally remained unchanged.[143,148] The IL-2 response tends to diminish with age, accounting for some of the decrease in cell proliferation in response to mitogenic and antigenic stimulation.
Rall et al.[149] compared the cytokine levels and responses of healthy elderly individuals (aged 65 to 80 years) with those of young individuals. Blood isolated from resting, unstimulated elderly individuals had a 50% greater TNFα response to Streptococcus epidermis than did that of young individuals. However, the response to lipopolysaccharide was unchanged. Lighart et al.[150] determined that, compared with young individuals, the elderly (mean age 74 years) had significantly elevated IL-6, TNFα and IL-1β levels in the supernatants of mitogen-stimulated cultures. Shinkai et al.[143] showed that blood taken from healthy elderly individuals had normal IL-1β, IL-4 and IFNγ responses to in vitro stimulation, but IL-2 production was impaired in comparison to young individuals.
Can training reverse the depression of immune function associated with age? Rall et al.[149] investigated the effects of 12 weeks of strength training on cytokine responses in the young, the elderly and patients with rheumatoid arthritis. Twice weekly exercise bouts comprised 3 sets of 8 repetitions of 1 repetition maximum (RM) contractions. There were no changes in IL-1β, TNFα, IL-6, IL-2 or prostaglandin production. However, the duration of the stimulus for adaptation as well as the exercise modality employed may have been inappropriate to induce the desired changes. Several other studies have investigated the difference in T cell function between sedentary and aerobically conditioned elderly individuals.[140] Both NK cell activity (measured in lytic units) and lymphocyte proliferation in response to PHA have generally been higher among highly conditioned individuals. These studies have used aerobically trained rather than strength-trained athletes. Shinkai et al.[143] compared the resting immune function of 17 elderly male runners (mean age 63.8 years) with 16 young (mean age 23.6 years) and 19 sedentary elderly (mean age 65.8 years) individuals. The active elderly individuals had an average aerobic power that was 33% greater than that of sedentary controls. They retained superior T cell proliferative responses to pokeweed mitogen and PHA, as well as a stronger production of IL-2, IFNγ and IL-4. Animal studies generally support these observations. Hoffman-Goetz and Sweeny[151] determined the effects of 8 weeks of voluntary wheel running in female, outbred Swiss Webster mice. NK/LAK activity against YAC-1 tumour cell targets was significantly higher in physically active animals compared with those not given access to a running wheel. However, it remained unclear whether the increased cytolytic activity was attributable to increased IL-1 or IL-2 production by the active animals. Nasrullah and Mazzeo[125] demonstrated that 15 weeks of aerobic training improved the T cell proliferative response of elderly rats, and augmented their ability to produce IL-2.
These and other animal and human studies suggest that regular exercise may slow age-related changes in cytokine production. IL-2 production is impaired with age, whereas production of IL-3 and IL-4 remains the same or may even increase.[148] With advancing age, there seems to be a relative expansion of the TH2 subset of T cells at the expense of TH1 cells. The data generated thus far lead to the conclusion that exercise has the potential to decelerate the age-related decline in cytotoxic cellular immune function. It will be interesting to discover whether other age-linked changes in cellular and soluble parameters are affected by training and are related to the individual’s level of fitness.
5. Physical Activity as a Model of Trauma and Sepsis
Since the physiological response to exercise shows many similarities to the changes observed during trauma and infection, exercise has been proposed as a model of sepsis and trauma.[1] The cytokine responses to clinical and experimental sepsis, trauma and thermal injury have been well characterised,[24,152–155] and they can be contrasted with the response to physical activity.[156] Discussion of the exercise model of generalised inflammation must examine qualitative and quantitative similarities and differences relative to other types of stimuli. Kinetic data on cytokine modulation must also be scrutinised, as timing of the induction of some cytokines influences the ability of the body to recover from their onslaught. Furthermore, data on the source of cytokines must be synthesised in the hope of developing endogenous and exogenous intervention and modulation strategies. Finally, the holistic consequences of the cytokine response must be compared for the exercise stimulus and sepsis/trauma: host response to changes in baseline values is ultimately more important than the degree of change in these values, as mechanisms exist by which the body can defend itself against its own cytokines.
5.1 Tissue Damage
Tissue damage is an obvious consequence of trauma and thermal injury, and may be caused indirectly by infection and sepsis.[157] Forced lengthening or eccentric exercise causes tissue damage during physical activity (table IV). Hesselink et al.[172] found that when isolated muscle was subjected to 120 or more eccentric contractions, muscle damage could be observed relative to isometrically contracted or control muscle tissue. In this study,[172] muscle damage was defined as an infiltration of fibres with mononuclear cells, the formation of multiple nuclei, or obvious swelling on histological examination. Clarkson et al.[160] proposed that the immediate loss of strength associated with eccentric exercise may be caused by the overstretching of sarcomeres. A decrease in overlap between actin and myosin filaments reduces sustainable contractile force and predisposes to damage. In support of this hypothesis, Newham and Ghosh[173] found that strength losses were greater if eccentric exercise was performed close to the extended extremity of the range of a muscle than if it was performed near to its contracted extremity. Armstrong et al.[158] reported that disruptions in the striation patterns of slow twitch rat extensor muscles were seen immediately following eccentric running.

Direct and latent/indirect tissue damage associated with physical activity, trauma and sepsis
At the molecular level, thiol proteases such as calpain mediate protein degradation of muscle myofibrillar complexes, with depletion of sarcomeric actinin which is an integral constituent of the Z-line.[163] MacIntyre et al.[174] suggested that activation of such proteases during eccentric activity may result in Z-line streaming and disorganisation of the normal alignment of myofilaments. Newham and Ghosh,[173] showed that exercise-induced disruption of the Z-line may persist over several days. Bruunsgaard et al.[78] compared concentric and eccentric cycle ergometry; tissue damage indicated by an increase in circulatory creatine kinase was observed after the forced-lengthening activity.
Although eccentric exercise induces tissue damage, it may be qualitatively different from that induced by trauma and thermal injury. Only contractile, connective and skeletal tissues are involved, and the lesions are generally microscopic and evenly distributed rather than focal. Unlike other injuries, repair can be effected in as little as 48 hours.[174] Physical activity-induced tissue damage, furthermore, may be a necessary preliminary for adaptive remodelling of the musculo-skeletal tissues.
Indirect tissue damage occurs as a consequence of thermal injury, mechanical injury and sepsis. Infiltration by activated immune cells is positively correlated with the extent of tissue damage, and some studies have demonstrated increased phagocytic cell oxidative activity in response to short term exercise.[1] Vindenes et al.[157] found that despite up-regulation of the chemokine IL-8, thermal injury did not change mouse phagocyte function, as indicated by the migration of cells from the circulation into the tissues. However, Cetinkale et al.[167] found increased malondialdehyde levels in blood taken 24 hours after severe thermal injury, indicating increased oxidative stress. The authors concluded that both direct thermal injury and immune cell oxidative processes contributed to increased lipid peroxidation. Battal et al.[169] studied the blocking of endothelin-A and -B activity during thermal injury. Endothelins potentiate the effect of local thrombosis to the point of occluding the microcirculation adjacent to a burn, causing necrotic damage of otherwise unaffected tissue. Exogenous antagonists to the endothelin receptors reduce the extent of tissue damage following burns.
Antioxidant status also has an important influence on the healing of thermal injuries, indicating the contribution of oxidative stress to tissue damage during this type of physical insult.[175] Major surgical interventions also cause vast tissue disruption, but macrophage and neutrophil activation causes less secondary tissue microtrauma than in the case of exercise. Infection often exacerbates tissue damage following thermal injury or surgery, triggering the action of innate and adaptive immune cells.[176] In the absence of infection, innate immune cell function is directed toward restoration of normal function in the vicinity of injury. Phagocytosis and coagulatory mechanisms clean up detritus and sterilise the area, this stage being followed by angiogenesis and functional restoration of native tissue.[138] Neutrophils and monocytes/macrophages play an important role. Neutrophils normally have a circulatory life span of approximately 6 days, but during sepsis they are rescued from apoptosis by cytokines such as TNFα and IL-6, as well as by circulatory indicators of sepsis such as lipopolysaccharide.[164] The survival of monocytes in human blood is also increased by lipopolysaccharide, but the more differentiated macrophages show a fragmentation of genomic DNA. This indicates apoptosis, and it makes data on these cell populations difficult to interpret in relation to the tissue damage induced by sepsis.[164] Bliff et al.[165] speculated that the increased lifespan of monocytes, coupled with a greater chemokine stimulus for extravasation, could cause greater exposure to activated immune cells and potentially more oxidative damage of otherwise healthy tissue.
Recent research has begun to elucidate the roles of NO, a product of activated immune cells, in sepsis and infection. Although adequate levels of NO production are necessary to preserve perfusion and carry out cytoprotective functions, overproduction contributes to haemodynamic instability and tissue damage.[166] NO is the main mediator of vasodilatation and the hyperdynamic circulation seen in sepsis, which along with tissue damage, causes multiple organ failure. The restoration of perfusion pressure is thus a major therapeutic goal in preventing further tissue damage. The TNFα produced by monocytes and macrophages in response to microbial products, including endotoxin, is a potent primer of neutrophil oxidative activity.
Evidence that heavy physical activity also causes immune cell-induced tissue damage is at least as conclusive. Table IV summarises the direct and latent tissue damage induced by physical activity, trauma and sepsis. The infiltration of muscle tissue by phagocytic cells during and after exercise may serve an adaptive and restorative role, rather than fostering an inappropriate immune autoreaction. In some models such as ischaemia/reperfusion, neutrophils can damage muscle tissue directly, but evidence of such damage in muscle tissue after physical activity is still inconclusive.[174] Although macrophages are present in human skeletal muscle both at rest and during exercise, their function is as yet unknown. To establish conclusively whether local phagocytic cells are responsible for the latent tissue damage observed after exercise, it will be necessary to compare intramuscular phagocyte function before and after exercise. It is possible that macrophages mainly mediate muscle rebuilding and hypertrophy.[174] Their presence seems a requirement for regeneration of skeletal muscle tissue; specialised macrophage subpopulations infiltrate muscle tissue during different stages of the recovery process.[161]
5.2 Comparison of the Cytokine Response to Exercise and to Inflammation
Although eccentric exercise is the primary mode of physical activity that induces detectable tissue damage, it may not be the most representative of the systemic and cytokine responses to physical injury or sepsis. Table V describes the cytokine response to various pathological inflammatory stimuli.

Plasma or peripheral blood mononuclear cell (PBMC) cytokine response to physical trauma, thermal injury and septic infection
Several studies have evaluated the appropriateness of exercise-induced muscle damage as a model of inflammation.[1] Bruunsgaard et al.[78] studied the cytokine response to 30 minutes of eccentric (resisted) cycle ergometry, finding that muscle damage was required to increase plasma IL-6 levels; neither injury nor increased IL-6 were seen in concentric exercise. However, creatine kinase levels did not peak until well after the appearance of IL-6, indicating that IL-6 is more rapidly induced than the usually accepted indicator of muscle damage, creatine kinase. Cannon and colleagues[86] found that IL-1β protein was elevated in the muscle tissue of individuals who performed 45 minutes of downhill running, although samples were evaluated for relative IL-1β staining intensity only 45 minutes and 5 days after exercise, and no data were obtained on the kinetics of IL-1β production in between these time points. Fielding et al.[84] used a comparable protocol to evaluate the immunohistochemical staining of intramuscular IL-1β. They found a 135% increase immediately after exercise and a 250% increase 5 days after exercise. The handful of experiments investigating the cytokine response to eccentric exercise point to a promising model of tissue damage and inflammation, but more work is needed to evaluate the efficacy of this mode of exercise in mimicking the responses observed in subclinical inflammation.
Endotoxin may be a trigger of the cytokine response to physical activity. Heat stroke-induced ischaemia of the gut induces release of lipopolysaccharide into the blood in rats.[195] Rosenberg et al.[196] detected increased lipopolysaccharide in the blood of athletes after a triathlon competition. Brock Utne et al.[197] found that plasma lipopolysaccharide and anti-lipopolysaccharide antibody levels were elevated after a long endurance run. It is possible that exercise-induced ischaemia of the gastrointestinal system increases gut permeability sufficiently to allow penetration by bacterial toxins, with a release of endogenous endotoxin, thus triggering the inflammatory response via immune cell activation.
Prolonged moderate or exhaustive exercise has shown promise as a model of cellular and cytokine inflammatory responses. Ostrowski et al.[85] demonstrated that the strenuous exercise-induced circulatory release of cytokines is sequentially similar to that induced during subclinical inflammation and sepsis. In marked similarity to both lethal and sublethal models of sepsis, TNFα was the first cytokine to be released, followed in sequence by IL-6, IL-1β and IL-1ra. Importantly, the 100-fold elevation of IL-6 with exercise was quantitatively similar to the circulatory increases observed during sepsis. However, the modest (2.5-fold) increases in IL-1β and TNFα, and difficulties in reproducing competitive stress levels in the laboratory, limit the applicability of this protocol as a model for inflammation. Detailed kinetic studies are needed to determine whether the sequential release of cytokines in response to physical activity bears similarity to the well characterised response to sepsis.
Brenner et al.[76] compared the cytokine and immune cell responses to resistance training, short intense exercise and prolonged endurance-type activity. Although creatine kinase was significantly elevated only after resistance exercise, the cytokine release profile indicated that prolonged endurance activity (in this case 2 hours of cycle ergometry at 60 to 65% V̇O2max) was most closely representative of the subclinical inflammatory response. In this study, prolonged exercise induced a transient rise in IL-6 that peaked 3 hours after exercise, but circulating TNFα levels increased slowly for 72 hours and may still have been climbing at the last sampling point. The modest extent of the exercise-induced increase in cytokine concentration brings into question the physiological significance of such slight responses. In this study, modulation of IL-10 was observed only after brief, intense exercise, in response to which circulatory IL-10 was reduced slightly but significantly. Therefore, the induction of tissue damage may not be of primary importance in using exercise as a model of the inflammatory response.
Unfortunately, there has been very little research on the effect of various forms of trauma and sepsis on cytokines essential for the cell-mediated response (for instance, IL-2 and IFNγ). The task of evaluating exercise models of inflammation is further impeded by a lack of solid characterisation of the cytokine response to the various types of physical activity. Characterisation of the effects of exercise on newly emerging cytokines may help define similarities and differences of cytokine response to the 2 types of stimuli.
Despite present limitations, it is meaningful to contrast kinetically, qualitatively, quantitatively and symptomatologically the cytokine response to prolonged physical activity with that induced by trauma or infection. Cytokine responses to lethal and sublethal sepsis begin with the rapid circulatory appearance of TNFα; within 60 minutes, concentrations increase many hundred-fold.[152] This response is followed closely by that of IL-6, which may increase many thousand-fold.[152,189,198] Circulatory IL-1β rises in response to infection, with the extent of the response depending on the severity of infection. It may be up-regulated by TNFα, whereas the chemokine IL-8 seems to follow the kinetics of IL-6.[152,189] In severe cases, the anti-inflammatory cytokines such as IL-1ra and IL-10 have negligible physiological significance compared with the onslaught of the pro-inflammatory mediators.[152] For example, in a murine lethal endotoxin challenge model, Napolitano et al.[199] demonstrated that circulatory IL-4 was only up-regulated if exogenous anti-IL-10 monoclonal antibodies were also administered; this indicates a possible interdependency of these cytokines.
The cytokine response to major mechanical trauma is consistently characterised by a transient elevation of IL-6. The regulation of other pro-inflammatory cytokines tends to be stimulus-specific, and there is often a global suppression of cytokine production.[200] One consistent finding is the circulatory up-regulation of immunosuppressive and anti-inflammatory cytokines such as IL-10.[138] Tissue damage, both in the context of exercise and with deliberate physical trauma, appears to up-regulate IL-6 production, suggesting that eccentric exercise may provide a particularly useful model of the cytokine response to direct physical trauma and inflammation. In developing this model, it will be instructive to determine the effect of eccentric activity on the anti-inflammatory cytokines.
Symptomatologically, the disease state correlates well with transient peaks of the pro-inflammatory cytokines in most forms of inflammation. Depending on the magnitude of response, the inflammatory state is either resolved or ends terminally. Thermal injury and physical trauma are more difficult to characterise because of the incomparability of implemented experimental designs. The pro-inflammatory cytokines seem to be up-regulated by trauma, but significant immunosuppression is also induced by other soluble mediators such as IL-4 and IL-10.[177] Although the cytokine response to heavy physical activity superficially resembles that accompanying trauma and infection, mechanisms may be very different, and typically there are marked differences in symptomatology. In most cases the cytokine response even to sublethal sepsis differs by several orders of magnitude, making it difficult to use one condition as a model of the other. Even the heaviest forms of physical activity induce only mild cytokine alterations when compared with sepsis. Moreover, ethical considerations preclude the implementation (in humans) of protocols that are punishing enough to elicit significant tissue damage. Distinctions abound: for example, sepsis depresses myocardial function, whereas physical activity usually does not.[122,201] However, physical activity induces endogenous pyrogenic activity in human blood, implying that changes in cytokine levels or their reception mimic, to some degree, the changes observed in inflammation.[202]
Tissue damage induced by exercise is repaired within a few days and usually results in a positive adaptation to the stimulus. Immune cells responsible for cytokine production play mainly a restorative role after exercise, whereas after physical trauma and infection, innate and specific immune cells exhibit a more aggressive response.[174] Differences in cytokine profiles induced by these various forms of trauma indicate that even the most severe levels of physical activity offer at best a scaled model of the inflammatory response in pathological states. The response to exercise is not entirely comparable to the immune activation observed during physical trauma and infection. However, both short term exercise and training affect immune function and effect immune adaptation, so that exercise immunology is important in its own right. The handful of experiments on training and immune function performed to date indicate that exercise may have a beneficial impact on cellular immune function, particularly in the elderly.
6. Conclusions and Future Prospects
Physical activity can modulate cytokine production from the level of gene expression to protein ligand and receptor release, with associated local and systemic consequences. Since cytokines are intimately involved in regulating immune function, studying the effects of exercise on cytokine production is warranted and relevant. Exercise immunologists must examine the effects of different types of short term exercise on cytokine production to understand the immediate implications for immunity. Physical activity should range from a low intensity to stressful competition. Adaptation to habitual physical activity seems to affect immune function, particularly cellular immunity, and this effect must be investigated more fully, using well controlled, longitudinal studies.
Exercise immunologists have traditionally looked at well known cytokines, including the pro-inflammatory mediators, some anti-inflammatory cytokines, a single chemokine, the IL-2 growth factor and the interferons. Very little attention has been directed to the haemopoietic cytokines, such as IL-3, the c-kit ligand [stem cell factor (SCF)] IL-5, or IL-7 and IL-14, the B cell growth and differentiation factors.[137,203] Investigation of IL-5 may be important, considering its ability to regulate eosinophil function.[204,205] The elusive pleiotropic growth factor IL-9 has also received very little attention by exercise immunologists.[206] The effects of short and long term exercise on newly discovered cytokines require elucidation, and findings need to be contrasted with the modulation of these soluble mediators by other inducers of inflammation. Provocative results on exercise modulation of IL-2 and its receptor dictate the need for further study of its close functional relative, IL-15, considering both the overlap of properties with IL-2, and its resistance to down-regulation by IL-4, IL-13, transforming growth factor β and IL-10.[58] Regulation of the newly characterised chemokine IL-16 and other well known chemokines such as RANTES and macrophage inflammatory protein (MIP) need investigation in the context of physical activity and the general inflammatory response.[48,60,207] The responses to exercise, trauma and infection of the T cell-derived IL-17, which induces the production of pro-inflammatory and haemopoietic cytokines, as well as the IFNγ-inducing factor, IL-18, may also be revealing.[208–210] LIF, a pleiotropic cytokine with seemingly unrelated effects on several cell types such as adipocytes, hepatocytes, and neural cells, should also be investigated in the context of inflammation.[211] Unfortunately, efforts to quantify the response of some of the ‘newer’ cytokines will be limited to laboratories capable of designing quantification protocols de novo, as, to date, there are no commercially available reagents with which to pursue this endeavour.
Modulation of cytokine gene expression has been explored extensively in other fields, but has had only limited examination in exercise physiology or generalised inflammation research.[87,97,137,212] To interpret data on the kinetics of cytokine gene expression meaningfully in the light of intra- and extracellular protein production and release, it will be necessary to understand in which cases protein synthesis and release is secondary to gene expression. For example, keratinocytes stimulated topically with a derivative of the immune modulator imiquimod, a possible antitumor and antiviral agent, up-regulate gene expression of IL-1α, IL-8, TNF and IFNγ.[213] Cytokine protein production by these cells increases predictably for all but IFNγ, the synthesis of which actually decreases.
Quantification of gene expression may be meaningless without consideration of the internal environment of the affected cell. In quantifying gene expression, it is necessary to establish not only the change in transcript level, but also the level of transcript stability. The latter depends on post-transcriptional modification of the nascent RNA by processes such as capping, methylation and polyadenylation, as well as levels and reactivities of endogenous ribonucleases.[5] Kinetic studies exploring the relationship of cytokine gene expression to circulatory protein levels have to be established for a variety of inflammatory stimuli, including physical activity, trauma and infection.
The cytokine response to physical activity deserves close inspection, because it is the manner in which the body responds to these small, soluble messengers rather than the severity of the insult itself that dictates symptomatology and clinical outcome.[1] The cytokines merit investigation because, unlike soluble endocrine mediators, they generally exhibit biological potency in the nano- to picomolar concentration range, and they have a potential for powerful modulation by stimuli such as infection and exercise. Similarities in cytokine response to infection and exercise have focused on the appropriateness of physical activity as a model for various forms of injury and inducers of inflammation. Future research should determine how far these striking similarities hold under close scrutiny. Resemblances may turn out to be superficial and limited to the cytokines studied thus far. Considering the uniqueness of most biological response equilibria, and the differences in pathophysiology and outcome between exercise-induced and other forms of inflammation, it seems likely that future studies will emphasise the distinctiveness of the cytokine response to physical activity. There are clearly qualitative differences in the way the body responds to exercise and the way it reacts to trauma and infection. However, this is not surprising, considering the difference in type and magnitude of the respective stimuli.
References
Shephard RJ, Shek PN. Physical activity and immune changes: a potential model of subclinical inflammation and sepsis. Crit Rev Rehab Phys Med 1996; 8 (3): 153–81
Rhind SG, Shek PN, Shephard RJ. The impact of exercise on cytokines and receptor expression. Exerc Immunol Rev 1995; 1: 97–148
Northoff H, Enkel S, Weinstock C. Exercise, injury and immune function. Exerc Immunol Rev 1995; 1: 1–25
Shephard RJ, Shek PN. Immune responses to inflammation and trauma: a physical training model. Can J Physiol Pharmacol 1998; 76: 469–72
Kuby J. Cytokines. In: Immunology. 2nd ed. New York: W.H. Freeman and Co., 1994: 297–322
Dinarello CA. The biological properties of interleukin-1. Eur Cytokine Netw 1994; 5 (6): 517–31
Kostura MJ, Tocci MJ, Limjuco G, et al. Identification of monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA 1989; 86: 5227–31
Lennard AC. Interleukin-1 receptor antagonist. Crit Rev Immunol 1995; 15: 77–105
Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med 1988; 167: 1708–12
Beasley D, Cohen RA, Levinsky NG. Interleukin 1 inhibits contraction of vascular smooth muscle. J Clin Invest 1989; 83: 331–5
Beasley D, Schwartz JH, Brenner BM. Interleukin-1 induces prolonged L-arginine-dependent cyclic guanosine monophosphate and nitrite production in rat vascular smooth muscle cells. J Clin Invest 1991; 87: 602–8
Gulick T, Chung MK, Pieper SJ, et al. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci USA 1989; 86: 6753–7
Coceani F, Lees J, Dinarello CA. Occurrence of interleukin 1 in cerebrospinal fluid of the conscious cat. Brain Res 1988; 446: 245–50
Konig A, Muhlbauer RC, Fleisch H. Tumor necrosis factor and interleukin 1 stimulate bone resorption in vivo as measured by [3-H] tetracycline excretion from prelabeled mice. J Bone Miner Res 1988; 3: 621–7
Zamir O, Hasselgren PO, O’Brien W et al. Muscle protein breakdown during endotoxemia in rats before and after treatment with interleukin-1 receptor antagonist (IL-1ra). Ann Surg 1992; 216: 381–5
Fibbe WE, Falkenburn JHF. Regulation of hematopoiesis by interleukin-1. Biotherapy 1990; 2: 325–35
Simic MM, Stocic-Grujicic S. The dual role of interleukin-1 in lectin-induced proliferation of T cells. Folia Biol 1985; 31: 410–24
Dejana E, Bertocchi F, Bortolami MC, et al. Interleukin-1 promotes tumor cell adhesion to cultured human endothelial cells. J Clin Invest 1988; 82: 1466–70
Coceani F, Lees J, Reford J, et al. Interleukin-1 receptor antagonist: effectiveness against interleukin-1 fever. J Physiol Pharmacol 1992; 7: 1590–6
Kammuler ME. Recombinant human interleukin-6: safety issues of a pleiotropic growth factor. Toxicology 1995; 105: 91–107
Ndubuisi MI, Patel K, Rayanade RJ, et al. Distinct classes of chaperoned IL-6 in human blood: differential immunological and biological activity. J Immunol 1998; 160: 494–501
Simpson RJ, Hammacher A, Smith DK, et al. Interleukin-6: structure-function relationships. Protein Sci 1997; 6: 929–55
Cohen MC, Cohen S. Cytokine function: a study in biologic diversity. Am J Clin Pathol 1996; 105: 589–98
Baumann H, Gauldie J. The acute phase response. Immunol Today 1994; 15: 74–80
Crane LJ, Miller DL. Plasma protein induction by isolated hepatocytes. Mol Cell Biochem 1983; 53/54: 89–109
Asano S, Okano A, Ozawa K. In vivo effects of recombinant interleukin-6 in primates: stimulated production of platelets. Blood 1990; 75: 1602–5
Hirano T. Interleukin-6 and its relation to inflammation and disease. Clin Immunol Immunopathol 1992; 62: 60–5
Hirano T, Akira S, Taga T, et al. Biological and clinical aspects of interleukin 6. Immunol Today 11: 443–8
Linker-Israeli M, Deans RJ, Wallace RJ, et al. Elevated levels of endogenous IL-6 in systemic lupus erythromatosus: a putative role in pathogenesis. J Immunol 1991; 147: 117–23
Tilg H, Trehu E, Atkins MB, et al. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor p55. Blood 1994; 83: 113–8
Hirano T, Matsuda T, Nakajima K. Signal transduction through gp130 that is shared among the receptors for the interleukin 6 related cytokine subfamily. Stem Cells 1994; 12: 262–77
Pennica D, Nedwin GE, Hayflick JS. Human tumor necrosis factor: precursor structure, expression, and homology to lymphotoxin. Nature 1984; 312: 724–9
Bemelmans MHA, van Tits LJH, Buurman WA. Tumor necrosis factor: function, release and clearance. Crit Rev Immunol 1996; 16: 1–11
Rowsey PJ, Borer KT, Kluger MJ. Tumor necrosis factor is not involved in exercise-induced elevation in core temperature. Am J Physiol 1993; 265 (6 Pt 1): R1351–4
Long NC, Vander AJ, Kunkel SL, et al. Antiserum against tumor necrosis factor increases stress hyperthermia in rats. Am J Physiol 1990; 258 (3 Pt 2): R591–5
Bender BA, Milsark IA, Cerami A. Cachectin/tumor necrosis factor: production, distribution, and metabolic fate. J Immunol 1985; 135: 3972–7
Aderka D, Engelmann H, Maor Y, et al. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med 1992; 175: 323–9
Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol 1993; 11: 571–611
Derynck R, Leung D, Gray P, et al. Human interferon is encoded by a single class of mRNA. Nucleic Acids Res 1982; 10: 3605–15
Billiau A. Interferon gamma: biology and role in pathogenesis. Adv Immunol 1996; 62: 61–130
Mantovani A, Dejana E. Cytokines as communication signals between leukocytes and endothelial cells. Immunol Today 1989; 10: 370–5
Ortaldo JR. Regulation of natural killer activity. Cancer Metastasis Rev 1987; 6: 637–51
Reiter Z. Interferon: a major regulator of natural killer cell-mediated cytotoxicity. J Interferon Res 1993; 13: 247–57
Young HA. Regulation of interferon-gamma gene expression. J Interferon Res 1996; 16: 563–8
Ruggiero V, Tavernier J, Fiers W, et al. Induction of synthesis of tumor necrosis factor alpha by interferon gamma. J Immunol 1986; 136: 2445–50
Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell 1992; 70: 705–7
Gresser I. Biological effects of interferons. J Invest Dermatol 1990; 95: 66S-71S
Gilman-Sachs A, DuChateau B. Clinical relevance of chemokines [abstract]. Clin Immunol Newsl 1997; 17 (7): 93
Malkovsky M, Sondel PM. Interleukin 2 and its receptor: structure, function and therapeutic potential. Blood Rev 1987; 1: 254–66
Lai KN, Leung JCK, Lai FM. Soluble IL-2 receptor release, interleukin 2 production, and interleukin 2 receptor expression in activated T -lymphocytes. Pathology 1991; 23: 224–8
Rubin LA, Nelson DL. The soluble interleukin-2 receptor: biology, function, and clinical application. Ann Intern Med 1990; 113: 619–27
Taga K, Kasahara Y, Yachie A, et al. Preferential expression of IL-2 receptor subunits on memory populations within CD4+ and CD8+T cells. Immunology 1991; 72: 15–9
Sheldon A, Flego L, Zola H. Coexpression of IL-2 receptor p55 and p75 by circulating blood lymphocytes. J Leuk Biol 1993; 54: 161–6
Aribia M-HB, Moire N, Metivier D, et al. IL-2 receptors on circulating natural killer cells and T lymphocytes. J Immunol 1989; 142: 490–9
Nagler A, Lanier LL, Phillips JH. Constitutive expression of high affinity interleukin 2 receptors on human CD 16-natural killer cells in vivo. J Exp Med 1990; 171: 1527–33
Scheibenbogen C, Keilholtz U, Richter M, et al. The interleukin-2 receptor in human monocytes and macrophages: regulation of expression and release of the α and β chains (p55 and p75). Res Immunol 1992; 143: 33–7
Tsudo M, Uchiyama T, Uchino H. Expression of the Tac antigen on activated normal human B cells. J Exp Med 1984; 160: 612–7
Doherty TM, Seder RA, Sher A. Induction and regulation of IL-15 expression in murine macrophages. J Immunol 1996; 156:735–41
Giri JG, Ahdieh J, Eisenman K, et al. Utilization of the beta and gamma chains of the interleukin-2 (IL-2) receptor by the novel cytokine IL-15. EMBO J 1994; 13: 2822–30
Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol 1997; 15: 675–705
Greenberg MJ, Streiter RM, Kunkel SL, et al. Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J Infect Dis 1996; 173: 173–59
Strieter RM, Koch AE, Antony VB, et al. The immunopathology of chemotactic chemokines: the role of interleukin-8 and monocyte chemoattractant protein-1. J Lab Clin Med 1994; 123: 183–97
Gong J-H, Clark-Lewis I. Antagonists of monocyte chemoattractant protein-1 identified by modification of functionally critical NH2-terminal residues. J Exp Med 1995; 181: 631–40
Kluth DC, Rees AJ. Inhibiting inflammatory cytokines. Semin Nephrol 1996; 16 (6): 576–82
Brown MA, Hural J. Functions of IL-4 and control of its expression. Crit Rev Immunol 1997; 17: 1–32
Colotta F, Re F, Muzio M. Interleukin-13 (IL-13) induces expression and release of interleukin-1 (IL-1) decoy receptor in human polymorphonuclear cells. J Biol Chem 1994; 269: 12403–6
Te Velde A, Huijbens RJF, de Vries JE. IL-4 increases FcγRc membrane expression and FcγRc-mediated cytotoxic activity of human monocytes. J Immunol 1990; 144: 3046–51
de Vries JE, Zurawski G. Immunoregulatory properties of IL-13: its potential role in atopic disease. Int Arch Allergy Immunol 1995; 106: 175–9
Zurawski G, de Vries JE. Interleukin 13, an interleukin-4-like cytokine that acts on monocytes and B-cells, but not T-cells. Immunol Today 1994; 15: 19–26
Schindler R, Clark RD, Dinarello CA. Dissociation between interleukin-1β mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 1990; 265: 10232–7
Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor R, interleukin-4 and interleukin 10. Proc Natl Acad Sci 1993; 685: 713–39
Joyce DA, Gibbons DP, Green P. Two inhibitors of inflammatory cytokine release, interleukin 10 and interleukin 4, have contrasting effects on the release of soluble p75 tumor necro sis factor receptor by cultured monocytes. Eur J Immunol 1994; 24: 2699–705
Burdin N, Rousset F, Banchereau J. B-cell-derived IL-10: production and function. Methods 1997; 11: 98–111
Malefyt RW, Yssel H, Roncarolo M-G, et al. Interleukin-10. Curr Opin Immunol 1992; 4: 314–20
Pedersen BK, Ostrowski K, Rohde T, et al. The cytokine response to strenuous exercise. Can J Physiol Pharmacol 1998; 76: 505–11
Brenner IKM, Natale VM, Suntres ZE, et al. Impact of different types of exercise on components of the inflammatory response [abstract]. Med Sci Sports Exerc 1998; 30 (5 Suppl.): S 19
Brenner IKM, Natale VM, Vasiliou P, et al. Impact of three different types of exercise on components of the inflammatory response. Eur J Appl Physiol 1999; 80: 452–60
Bruunsgaard H, Galbo H, Halkjaer-Kristensen J, et al. Exercise-induced increase in serum interleukin-6 in humans is related to muscle damage. J Physiol (Lond) 1997; 499: 833–41
Nieman DC, Nehlsen-Cannarella SL, Fagoaga OR, et al. Influence of mode and carbohydrate on the cytokine response to heavy exertion. Med Sci Sports Exerc 1998; 30: 671–8
Gannon GA, Rhind SG, Slick PN, et al. Inhibition of exercise-induced cytokine release by thermal clamping [abstract]. FASEB J 2000; 14: A619
Moldoveanu AI, Shephard RJ, Slick PN. Prolonged exercise elevates plasma levels but not gene expression of IL-1 β, IL-6, and TNFα, in circulating mononuclear cells. J Appl Physiol 2000; 89: 1499–504
Rhind SG, Slick PN, Brenner IKM, et al. Intracellular and serum cytokine profiles after 8 days of exhaustive exercise and cold exposure [abstract]. FASEB J 2000; 14: A618
Haahr PM, Pedersen BK, Fomsgaard A, et al. Effect of physical exercise on in vitro production of interleukin 1, interleukin 6, tumor necrosis factor-α, interleukin 2 and interferon-gamma. Int J Sports Med 1991; 12: 223–7
Fielding R, Manfredi T, Ding W et al. Prolonged alterations in skeletal muscle ultrastructure following eccentric exercise. Clin Sci 1994; 87 Suppl.: 82–3
Ostrowski K, Rohde T, Asp S, et al. The sequential release of cytokines in strenuous exercise. Int J Sports Med 1998; 19 Suppl. 3: S216–7
Cannon JG, Fielding RA, Fiatarone MA, et al. Increased interleukin 1β in human skeletal muscle after exercise. Am J Physiol 1989; 257 (2 Pt 2): R451–5
Netea MG, Drenth JPH, De Bout N, et al. A semi-quantitative reverse transcriptase polymerase chain reaction method for measurement of mRNA for TNF-alpha and IL-1beta in whole blood cultures: its application in typhoid fever and eccentric exercise. Cytokine 1996; 8 (9): 739–44
Ullum H, Haahr PM, Diamant M, et al. Bicycle exercise enhances plasma IL-6 but does not change IL-1α, IL-6, or TNF-α pre-mRNA in BMNC. J Appl Physiol 1994; 77: 93–7
Ostrowski K, Rohde T, Zacho M, et al. Evidence that interleukin-6 is produced in human skeletal muscle during prolonged running. J Physiol (Lond) 1998; 508: 949–53
Natelson BH, Zhou X, Ottenweller JE, et al. Effect of acute exhausting exercise on cytokine gene expression in men. Int J Sports Med 1996; 17: 299–302
Anwar A, Smith LL, Holbert D, et al. Serum cytokines after strenuous exercise [abstract]. Med Sci Sports Exerc 1997; 29 Suppl.: S73
Lewicki R, Tchorzewski H, Majewska E, et al. Effect of maximal physical exercise on T-lymphocyte subpopulations and on interleukin 1 (IL 1) and interleukin 2 (IL 2) production in vivo. Int J Sports Med 1988; 9: 114–7
Suzuki K, Yamada M, Kurakake S, et al. Circulating cytokines and hormones with immunosuppressive but neutrophil-priming potentials rise after endurance exercise in humans. Eur J Appl Physiol 2000; 81: 281–7
Weinstock C, Konig D, Harnischmacher R, et al. Effect of exhaustive exercise on the cytokine response. Med Sci Sports Exerc 1997; 29: 345–54
Sprenger H, Jacobs C, Nain M, et al. Enhanced release of cytokines, interleukin-2 receptors and neopterin after long-distance running. Clin Immunol Immunopathol 1992; 63: 188–95
Bury TB, Louis R, Radennecker ME, et al. Blood mononuclear cells mobilization and cytokine secretion during prolonged exercises. Int J Sports Med 1996; 17: 156–60
Woods JA, Linner KM, Sharp BM. Effects of exhaustive exercise on LPS-induced IL-6 gene expression [abstract]. Med Sci Sports Exerc 1997; 26 (5 Suppl.): S 182
Nehlsen-Cannarella SL, Fagoaga OR, Nieman DC, et al. Carbohydrate and cytokine response to 2.5 h of running. J Appl Physiol 1997; 82: 1662–7
Papanicolaou DA, Pestrides JS, Tsigos C, et al. Exercise stimulates interleukin-6 secretion: inhibition by glucocorticoids and correlation with catecholamines. Am J Physiol 1996; 271 (3 Pt 1): E601–5
Rivier A, Pene J, Chanez P, et al. Release of cytokines by blood monocytes during strenuous exercise. Int J Sports Med 1994; 15: 192–8
Gannon GA, Rhind SG, Suzui M, et al. Changes in selected cellular and soluble mediators of immunity following a 250.5 km competitive road-cycling race. Proceedings, International Society of Exercise Immunology; 1995 Nov; Brussels, 40
Gannon GA, Rhind SG, Suzui M, et al. Circulating levels of peripheral blood leucocytes and cytokines following competitive cycling. Can J Appl Physiol 1997; 22: 133–47
Northoff H, Berg A. Immunologic mediators as parameters of the reaction to strenuous exercise. Int J Sports Med 1991; 12: 9–15
Suzuki K, Totsuka M, Nakaji M, et al. Endurance exercise causes interactions among stress hormones, cytokines, neutrophil dynamics, and muscle damage. J Appl Physiol 1999; 87: 1360–7
Rokitzki L, Logemann E, Keul J. Interleukin-6, tumor necrosis factor-alpha and malon-dialdehyde serum concentration during a marathon-run. Int J Sports Med 1994; 15: 360
Dufaux B, Order U. Plasma elastase-αl-antitrypsin, neopterin, tumor necrosis factor, and soluble interleukin-2 receptor after prolonged exercise. Int J Sports Med 1989; 10: 434–8
Espersen GT, Elbaek A, Ernst E, et al. Effect of physical exercise on cytokines and lymphocyte subpopulations in human peripheral blood. APMIS 1990; 98: 395–400
Baum M, Muller-Steinhardt M, Liesen H, et al. Moderate and exhaustive endurance exercise influences the interferon-gamma levels in whole-blood culture supernatants. Eur J Appl Physiol 1997; 76: 165–9
Bagby GJ, Sawaya DE, Crouch LD, et al. Prior exercise suppresses the plasma tumor necrosis factor response to bacterial lipopolysaccharide. J Appl Physiol 1994; 77: 1542–7
Drenth JPH, van Uun SHM, van Demen M, et al. Endurance run increases circulating IL-6 and IL-1ra but downregulates ex vivo TNF-α and IL-1β production. J Appl Physiol 1995; 79: 1497–503
Drenth JPH, Krebbers RJM, Bijzet J, et al. Increased circulating cytokine receptors and ex vivo interleukin-1 receptor antagonist and interleukin-1beta but decreased tumor necrosis factor-alpha production after a 5-km run. Eur J Clin Invest 1998; 28: 866–72
Northoff H, Weinstock C, Berg A. The cytokine response to strenuous exercise. Int J Sports Med 1994; 15 Suppl. 3: S167–71
Ostrowski K, Rohde T, Asp S, et al. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol (Lond) 1999; 515: 287–91
Sutton BJ, Gould HJ. The human IgE network. Nature 1993; 366: 421–8
Moyna NM, Acker GR, Fulton JR, et al. Lymphocyte function and cytokine production during incremental exercise in active and sedentary males and females. Int J Sports Med 1996; 17: 585–91
Castell L, Poortmans JR, Leclercq R, et al. Some aspects of the acute phase response after a marathon race. Eur J Appl Physiol 1997; 75: 47–53
Smith JA, Telford RD, Baker MS, et al. Cytokine immunoreactivity in plasma does not change after moderate endurance exercise. J Appl Physiol 1992; 73: 1396–401
Camus G, Poortmans J, Nys M, et al. Mild endotoxemia and the inflammatory response induced by a marathon race. Clin Sci 1997; 92: 415–22
Northoff H, Flegel W, Männel DN, et al. Increased levels of interleukin-6 (IL-6) and/or IL-7 in sera of long-distance runners. In: Dinarello C, Kluger ML, Powanda MC, etal., editors. The physiological and pathological effects of cytokines. New York (NY): Wiley-Liss, Inc., 1990: 75–9
Fitzpatrick DR, Shirley KM, McDonald LE, et al. Distinct methylation of the interferon gamma (IFN-gamma) and interleukin 3 (IL-3) genes in newly activated primary CD8+ T lymphocytes: regional IFN-gamma promoter demethylation and mRNA expression are heritable in CD44(high)CD8+ T cells. J Exp Med 1998; 188: 103–17
Umland SP, Razac S, Shah H, et al. Interleukin-5 mRNA stability in human T cells is regulated differently than interleukin-2, interleukin-3, interleukin-4, granulocyte/macrophage colony stimulating factor, and interferon gamma. Am J Respir Cell Mol Biol 1998; 18: 631–42
Mann DL. Stress activated cytokines and the heart. Cytokine Growth Factor Rev 1996; 7: 341–54
Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev 1999; 79: 1–71
Rhind SG, Slick PN, Shinkai S, et al. Effects of moderate endurance exercise and training on in vitro lymphocyte proliferation, interleukin-2 (IL-2) production, and IL-2 receptor expression. Eur J Appl Physiol 1996; 64: 348–60
Nasrullah I, Mazzeo RS. Age-related immunosenescence in Fischer 344 rats: influence of exercise training. J Appl Physiol 1992;73:1932–8
Gmünder FK, Joller PW, Joller-Jemelka HI, et al. Effect of a herbal yeast food supplement and long-distance running on immunological parameters. Br J Sports Med 1990; 24: 103–12
Fry RW, Morton AR, Crawford GMP, et al. Cell numbers and in-vitro responses of leukocytes and lymphocyte subpopulations following maximal exercise and interval training sessions of different intensities. Eur J Appl Physiol 1992; 64: 218–27
Tilz GP, Domej W, Diez-Ruiz A, et al. Increased immune activation during and after physical exercise. Immunobiology 1993; 188: 194–202
Gray AB, Smart YC, Telford RD, et al. Anaerobic exercise causes transient changes in leukocyte subsets and IL-2R expression. Med Sci Sports Exerc 1992; 24: 1332–8
Viti A, Muscatella M, Paulescu L, et al. Effect of exercise on plasma interferon levels. J Appl Physiol 1985; 59: 426–8
Smits HH, Grunberg K, Derijk RH, et al. Cytokine release and its modulation by dexamethasone in whole blood following exercise. Clin Exp Immunol 1998; 111 (2): 463–8
Surkina ID, Danilenko SV, Dudov NS, et al. The role of the immune system in processes of adaptations to stress in sportsmen. Clin Sci 1994; 87 Suppl.: 22
Nieman DC. Immune response to heavy exertion. J Appl Physiol 1997; 82: 1385–94
Nielsen HB, Hanel B, Loft S, et al. Restricted pulmonary diffusion capacity after exercise is not an ARDS-like injury. J Sports Sci 1995; 13: 109–13
Hopkins SR, Schoene RB, Henderson WR, et al. Intense exercise impairs the integrity of pulmonary blood-gas barrier in elite athletes. Am J Respir Crit Care Med 1997; 155 (3): 1090–4
Ostrowski K, Hermann C, Bangash A, et al. A trauma-like elevation in plasma cytokines in humans in response to treadmill running. J Physiol (Lond) 1998; 513: 889–94
Krishnaswamy G, Smith JK, Srikanth S, et al. Effect of moderate exercise on the TH1 and TH2 cytokine mRNA profiles of circulating lymphocytes. J Allergy Clin Immunol 1997; 95 (1 Pt 2): 359
Klava A, Windsor ACJ, Farmery SM, et al. Interleukin-10: A role in the development of postoperative immnunosuppression. Arch Surg 1997; 132: 425–9
Baum M, Klopping-Menke K, Muller-Steinhardt M, et al. Increased concentrations of interleukin-l-beta in whole blood culture supernatants after 12 weeks of moderate endurance exercise. Eur J Appl Physiol 1999; 79: 500–3
Nieman DC. Exercise, infection and immunity. Int J Sports Med 1994; 15 Suppl.: S131–41
Shephard RJ, Shek PN. Potential impact of physical activity and sport on the immune system: a brief review. Br J Sports Med 1994; 28: 247–55
Shephard RJ, Slick PN. Impact of physical activity and sport on the immune system. Rev Environ Health 1996; 11: 133–47
Shinkai S, Kohno H, Kimura K, et al. Physical activity and immune senescence in men. Med Sci Sports Exerc 1995; 27: 1516–26
Baj Z, Kantorski J, Majewska E, et al. Immune status of competitive cyclists before and after the training session. Int J Sports Med 1994; 15: 319–24
Weicher H, Werle E. Interactions between hormones and the immune system. Int J Sports Med 1991; 12 Suppl.: S30–7
Papa S, Vitale M, Mazzoti G, et al. Impaired lymphocyte stimulation induced by long-term training. Immunol Lett 1989; 22: 29–33
Rhind SG, Shek PN, Shinkai S, et al. Differential expression of interleukin-2 receptor alpha and beta chains in relation to natural killer cell subsets and aerobic fitness. Int J Sports Med 1994; 15: 911–8
Shinkai S, Konishi K, Shephard RJ. Aging, exercise, training and the immune system. Exerc Immunol Rev 1997; 3: 68–95
Rall LC, Roubenoff R, Cannon JG, et al. Effects of progressive resistance training on immune response in aging and chronic inflammation. Med Sci Sports Exerc 1996; 28: 1356–65
Lighart GJ, Corberand JX, Fournier C, et al. Admission criteria for immunogerontological studies in man: the SENIEUR protocol. Mech Ageing Dev 1990; 28: 47–55
Hoffman-Goetz L, Sweeny AL. Lymphokine activated killer cell activity following voluntary physical activity in mice. J Sports Med Phys Fitness 1994; 34: 83–90
Foex BA, Shelly MP. The cytokine response to critical illness. J Accid Emerg Med 1996; 13: 154–62
Kelly JL, O’Sullivan C, O’Riordain M, et al. Is circulating endotoxin the trigger for the systemic inflammatory response syndrome seen after injury. Ann Surg 1997; 225: 530–43
O’Sullivan S, Lederer JA, Horgan AF, et al. Major injury leads to predominance of the T helper-2 phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg 1995; 222: 482–92
van Zee KJ, Kohmo T, Fischer E, et al. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necro sis factor alpha in vitro and in vivo. Proc Natl Acad Sci USA 1992; 89: 4845–9
Shek PN, Sabiston BH, Buguet A, et al. Strenuous exercise and immunological changes. Int J Sports Med 1995; 16: 466–74
Vindenes H, Ulvestad E, Bjerknes R. Increased levels of circulating interleukin-8 in patients with large burns: relation to burn size and sepsis. J Trauma 1995; 39: 635–40
Armstrong RB, Ogilvie RW, Schwane JA. Eccentric exercise-induced injury to rat skeletal muscle. J Appl Physiol 1983; 54: 80–93
Pyne D. Regulation of neutrophil function during exercise. Sports Med 1994; 17: 245–58
Clarkson PM, Nosaka K, Braun M. Muscle function after exercise-induced muscle damage and rapid adaptation. Med Sci Sports Exerc 1992; 24: 512–20
St. Pierre B, Tidball JG. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J Appl Physiol 1994; 77: 290–7
Newham DJ. The consequences of eccentric contractions and their relationship to delayed onset muscle pain. Eur J Appl Physiol 1988; 57: 353–9
Reid W, Huang J, Byron S. Diaphragm injury and myofibrillar structure induced by resistive loading. J Appl Physiol 1994; 76: 176–84
Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock 1996; 6: S27–38
Bliff WL, Moore FA, Carl VS, et al. Interleukin-6 delays neutrophil apoptosis. Arch Surg 1996; 131: 24–9
Johnson ML, Billiar TR. The role of nitric oxide in surgical infection and sepsis. World J Surg 1998; 22: 187–96
Cetinkale O, Belce A, Konukoglu D, et al. Evaluation of lipid peroxidation and total antioxidant status in plasma of rats following thermal injury. Burns 1997; 23: 114–6
Sparkes BG. Influence of burn induced lipid-protein complex on IL-2 secretion by PBMC in vitro. Burns 1991; 17: 128–35
Battal MN, Hata Y, Matsuka K, et al. Reduction of progressive burn injury by using a new non-selective endothelin-A and endothelin-B receptor antagonist. Plast Recon Surg 1997; 99: 1610–9
Maxwell SR, Lip GR. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol 1997; 58: 91–117
Hack CE, Wolbink GJ, Schalkwijk C, et al. The role of secretory phospholipase A2 and C reactive protein in the removal of injured cells. Immunol Today 1997; 18: 111–5
Hesselink MKC, Kuipers H, Geurten P, et al. Structural muscle damage and muscle strength after incremental number of isometric and forced lengthening contractions. J Muscle Res Cell Motil 1996; 17: 335–41
Newham DJ, Ghosh G. Muscle fatigue and pain after eccentric contractions at long and short lengths. Clin Sci 1988; 74: 55–62
MacIntyre DL, Reid WD, McKenzie DC. Delayed muscle soreness. Sports Med 1995; 20: 24–40
Rock CL, Dechert RE, Khilnani R, et al. Carotenoids and antioxidant vitamins in patients after burn injury. J Burn Care Rehab 1997; 18 (3): 269–78
Leaper DJ. Risk factors for surgical infection. J Hosp Infect 1995; 30 Suppl.: 127–39
Kawakami M, Kaneko N, Anada H, et al. Measurement of interleukin-6, interleukin-10, and tumor necrosis factor-alpha levels in tissues and plasma after thermal injury in mice. Surgery 1997; 121: 440–8
Ohzato H, Monden M, Yoshizaki K. Systemic production of interleukin-6 following acute inflammation. Biochem Biophys Res Commun 1993; 197: 1556–62
Yamada Y, Endo S, Inada K. Plasma cytokine levels in patients with severe burn injury — with reference to the relationship between infection and prognosis. Burns 1996; 22: 587–93
O’Riordan DS, Mendez MV, O’Riordan MG. Molecular mechanisms of decreased IL-2 production after thermal injury. Surgery 1993; 114: 407–15
Schinkel C, Zimmer S, Kremer JP, et al. Comparative analysis of transcription and protein release of the inflammatory cytokines interleukin-1 beta (IL-1β) and interleukin-8 (IL-8) following major burn and mechanical trauma. Shock 1995; 4: 241–6
Marano MA, Fong Y, Moldawer LL. Serum cachectin and tumor necrosis factor in critically ill patients with burns correlates with infection and mortality. Surg Gynecol Obstet 170: 32–8
Gadd MA, Hansbrough JF, Hoyt DA, et al. Defective T-cell surface antigen expression after mitogen stimulation. Ann Surg 1989; 20: 112–8
Hariri RJ, Chang VA, Barie PS, et al. Traumatic injury induces interleukin-6 production by human astrocytes. Brain Res 1994; 636: 139–42
Kleinschmidt S, Wanner GA, Bubmann D, et al. Proinflammatory cytokine gene expression in whole blood from patients undergoing coronary bypass surgery and its modulation by pentoxifylline. Shock 1998; 9: 12–20
Martin C, Boisson C, Haccoun M, et al. Patterns of cytokine evolution (tumor necrosis factor and interleukin-6) after septic shock, hemorrhagic shock and severe trauma. Crit Care Med 1997; 25: 1813–9
Pirenne J, Mahieu X, de Groote D, et al. Effect of cardiac surgery on cytokine production. Cytokine Res 1993; 43: 13–8
Ayala A, Lehmann DL, Herdon CD, et al. Mechanism of enhanced susceptibility to sepsis following hemorrhage: interleukin-10 suppression of T cell response is mediated by eicosanoid-induced interleukin-4 release. Arch Surg 1994; 129: 1172–8
van der Poll T, Lowry SF. Tumor necrosis factor in sepsis: mediator of multiple organ failure of essential part of host response? Shock 1995; 3: 1–12
Michie HR, Manogue KR, Spriggs DR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med 1988; 318: 1481–6
Pruitt JH, Copeland EMI, Moldawer LL. Interleukin-1 and Interleukin-1 antagonism in sepsis, systemic inflammatory response syndrome, and septic shock. Shock 1995; 3: 235–51
Hesse DG, Tracey KJ, Fong Y, et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet 1988; 166: 147–53
Levi M, van der Poll T, Cate H, et al. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxernia. Eur J Clin Invest 1997; 27: 3–9
Fong Y, Tracey KJ, Moldawer LL, et al. Antibodies to cachectin/tumor necrosis factor reduce interleukin-1 beta and interleukin-6 appearance during lethal bacteremia. J Exp Med 1989; 170: 1627–33
DuBose DA, Basamania K, Maglione L, et al. Role of bacterial endotoxins of intestinal origin in rat heat stress mortality. J Appl Physiol 1983; 54: 31–6
Rosenberg AT, Brock Utne JG, Gaffin SL, et al. Strenuous exercise causes systemic endotoxemia. J Appl Physiol 1988; 65: 106–8
Brock Utne JG, Gaffin SL, Wells MT, et al. Endotoxemia in exhausted runners after a long-distance race. S Aft Med J 1988; 73: 533–6
Stricter RM, Kunkel SL, Bone RC. Role of tumor necrosis factor-alpha in disease states and inflammation. Crit Care Med 1993; 21: S447–63
Napolitano L, Campbell C, Bass BL. Kinetics of splenocyte interleukin-4 production after injury and lethal endotoxic challenge. J Surg Res 1997; 67: 33–9
Scotte M, Masson S, Lyoumi S, et al. Cytokine gene expression in liver following minor or major hepatectomy in rat. Cytokine 1997; 9: 859–67
Odeh M. Tumor necrosis factor-alpha as a myocardial depressant substance. Int J Cardiol 1993; 42: 231–8
Cannon JG, Kluger MJ. Endogenous pyrogen activity in human plasma after exercise. Science 1983; 220: 617–9
Ford R, Tamayo A, Martin B, et al. Identification of B -cell growth factors (interleukin 14; high molecular weight B-cell growth factors) in effusion fluids from patients with aggressive B-cell lymphomas. Blood 1995; 86: 283–93
Dent LA, Munro G, Piper KP, et al. Eosinophilic interleukin 5 (IL-5) transgenic mice: eosinophil activity and impaired clearance of Schistosoma mansoni. Parasite Immunol 1997; 19: 291–300
Hirai K, Miyamasu M, Takaishi T, et al. Regulation of the function of eosinophils and basophils. Crit Rev Immunol 1997; 17: 325–52
Gessner A, Blum H, Rollinghoff M. Differential regulation of IL-9 expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology 1993; 189: 419–35
Lim KG, Wan H-C, Bozza PT, et al. Human eosinophils elaborate the lymphocyte chemoattractants ILr16 (lymphocyte chemoattractant factor) and RANTES. J Immunol 1996; 156: 2566–70
Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 1996; 183: 2593–603
Stoll S, Muller G, Kurimoto M, et al. Production of IL-18 (IFN-gamma inducing factor) messenger RNA and functional protein by murine keratinocytes. J Immunol 1997; 159: 298–302
Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol 1995; 155: 5483–6
Kurzrock R, Estrov Z, Wetzler M, et al. LIF: not just a leukemia inhibitory factor. Endocrine Rev 1991; 12: 208–17
Rohde T, Ostrowski K, Zacho M, et al. Evidence that IL-6 is produced in skeletal muscle during intense long-term muscle activity. Eur J Physiol 1998; 78: 448–53
Fujisawa H, Shivji GM, Kondo H. Effect of a novel topical immunomodulatory, S-28463, on keratinocyte cytokine gene expression and production. J Interferon Res 1996; 16: 555–9
Acknowledgements
The research of Dr Shephard is supported in part by a grant from the Defence and Civil Institute of Environmental Medicine.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moldoveanu, A.I., Shephard, R.J. & Shek, P.N. The Cytokine Response to Physical Activity and Training. Sports Med 31, 115–144 (2001). https://doi.org/10.2165/00007256-200131020-00004
Published:
Issue Date:
DOI: https://doi.org/10.2165/00007256-200131020-00004