

The antifibrinolytic agent aprotinin, a naturally occurring serine protease inhibitor, was removed from the worldwide market in October 2007.1 Aprotinin and two other antifibrinolytic agents (the lysine analogues tranexamic acid and aminocaproic acid) had been used in cardiac surgery to minimize bleeding and reduce the need for transfusion. All three drugs have proven effective in reducing the need for blood transfusion as compared with placebo in clinical trials.2–5
The withdrawal of aprotinin was precipitated by the early termination of the Blood Conservation Using Antifibrinolytics in a Randomized Trial (BART).6 An interim analysis had shown an increase in deaths, from 4% to 6%, among patients at high risk who were undergoing cardiac surgery — a relative increase of 50% in patients receiving aprotinin compared with those receiving tranexamic or aminocaproic acid. However, in 2011, Bayer, the maker of aprotinin, was again allowed to market the drug in Canada and Europe. A Health Canada expert advisory panel and the European Medicines Agency stated that the BART and its primary report in the New England Journal of Medicine were flawed, and recent comments have supported that claim.7,8
As authors of the BART, we are concerned about the approach and decision-making in the process used by Health Canada to arrive at its 2011 decision, and about the subsequent rulings of Health Canada and the European Medicines Agency. We refute the three primary criticisms of the BART, and we believe that patients are being put at substantial risk in the presence of safer alternatives. Aprotinin remains unavailable for use in the United States.
The history of aprotinin research and its regulation
Several meta-analyses of placebo and open-label controlled trials between 1999 and 2005 suggested that aprotinin may decrease the risk of death, stroke and repeat surgery for massive bleeding compared with placebo.3,9,10 However, in 2006, concern arose over the safety of the drug when large observational studies noted an association between aprotinin and increased rates of cardiovascular and cerebrovascular complications, renal failure and short- and long-term mortality11–14 compared with other antifibrinolytic agents. Based on these risks, the United States Food and Drug Administration issued public advisories.15 A Cochrane review of 20 head-to-head comparisons published in 2005 suggested that there were still too few data to definitively recommend one drug over another.3
The BART16 was a large, pragmatic, double-blind trial that randomly allocated 2468 patients (2331 included in the primary analysis) undergoing cardiac surgical procedures using cardiopulmonary bypass to one of three antifibrinolytic treatments: tranexamic acid, aminocaproic acid or aprotinin. The BART aimed to include patients at high risk, which was defined as a twofold increase in risk of death and massive bleeding compared with routine isolated graft procedures. This twofold risk allowed us to characterize our population as “high-risk.” The trial included repeat grafts and combined graft procedures in which aprotinin was commonly used and approved in Canada. All drug dosages were based on a strategy of maximum effective dosing documented in clinical practice and in the literature.
In October 2007, the trial was terminated early by the BART Executive Committee on the advice of the Data Monitoring and Safety Board. The Board advised that patients no longer be enrolled in the aprotinin group owing to an increased risk of death. Massive bleeding rates tended to be lower among patients receiving aprotinin (9.5% v. 12.1% in the other groups; relative risk [RR] 0.79; 95% confidence interval [CI] 0.59–1.01). However, 30-day all-cause mortality was 6.0% in the aprotinin group, compared with 3.9% in the tranexamic acid group and 4.0% in the aminocaproic acid group (RR comparing aprotinin to both lysine analogues combined 1.53 [95% CI 1.06 to 2.20]). The strong and consistent negative mortality trend seen with aprotinin led the investigators to suggest that its use in high-risk cardiac surgery was unwarranted despite its potential for reducing massive bleeding.16 Shortly after the end of the trial, Bayer voluntarily withdrew aprotinin from the market.
In 2011, a Health Canada expert advisory panel17 and the European Medicines Agency18 both stated that the BART and its primary report in the New England Journal of Medicine were flawed, and these claims have been supported in recent comments.7,8 Bayer, the maker of aprotinin, was allowed to market the drug again in Canada and the European Union in 2011.
The reapproval of aprotinin
Health Canada, Canada’s drug regulator, convened an expert advisory panel to review the evidence for and against aprotinin in cardiac surgery. In their final report, the panel identified the 137 patients excluded from the final intention-to-treat analysis as their “most prominent issue.” They suggested that this threatened the validity of the trial’s findings. The panel noted that mortality trends in the excluded patients differed from those included in the final published analysis.
In addition, they noted that about 75% of massive bleeding events were reclassified from data provided to the Data Monitoring and Safety Board as compared with the final analysis.19 In addition to the advice and direction provided by the panel, Health Canada statisticians reproduced the primary and secondary analyses reported in the original publication using the original dataset and found them to be reproducible and accurate. Heath Canada then conducted a series of secondary analyses16 using the BART dataset. First, they explored the impact of severity of illness using the Toronto Risk Score. Second, they focused on the impact of heparin and protamine, noting several imbalances and missing information in the BART dataset.
Using internal analyses and the report prepared by the expert advisory panel, Health Canada recommended approval of aprotinin with warnings in September 2011, and requested that Bayer conduct further safety studies involving high-risk patients.17 In a press release explaining the decision, the panel suggested that the BART “was not designed to reliably determine the risk of death ... and that the increased number of deaths in Trasylol [aprotinin] patients could have been due to chance.” They went on to say that the drug could have led to increases in deaths due to blood clotting in patients receiving aprotinin if monitoring of clotting was inadequate during surgery.20
In February 2012, the European Medicines Agency Committee for Medicinal Products for Human Use also recommended that Bayer be allowed to resume marketing aprotinin. In its written rationale accompanying the report, the committee declared that “there were a number of problems with the way the BART study was conducted, which cast doubt on the previous conclusions.”18 Specifically, the committee referred to imbalances in the use of heparin and inappropriate monitoring of the use of anticoagulants, and again commented on the exclusion of patients from the primary analysis. The committee recommended allowing Bayer to market aprotinin with warnings and the establishment of a registry of patients who had received the drug.
Response to criticisms of the BART
We have identified three primary criticisms of the BART that facilitated aprotinin’s return to market: the reclassification of 75% of our primary outcomes, exclusion from our primary analysis of 137 patients after randomization, and baseline imbalances in the use of heparin and the monitoring of anticoagulation. We refute all three criticisms.
Reclassification of primary outcomes
Health Canada’s Expert Advisory Panel on Trasylol stated that the BART authors reclassified 75% of the primary outcome events.19 This estimate was incorrect. Despite our repeated attempts to view the reasoning behind this conclusion, the expert advisory panel provided no calculations or justification to BART investigators to support the estimate. Our true reclassification of primary outcomes was 1.6% (38/2438). During data cleaning and the verification of our case report forms, we identified 20 events in which the primary outcome was missed by study coordinators and site clinicians and 18 reported events that did not meet the definition of our primary outcome. Most of these 38 reclassifications were based on differences in the measurement of chest-tube blood loss over time and the adjudication of death due to hemorrhage. We notified Health Canada and the panel of our findings. As of September 2014, the panel has not provided an explanation for their claim of a 75% reclassification rate or acknowledged any error, and we have been left to speculate as to the reason.
During its deliberations, the panel requested that we rerun each original Data Safety and Monitoring Board report and compare them against verified final data to contrast inferences from the two datasets. In doing so, we provided the cumulative reclassified events on each report (i.e., reclassifications on each report would be carried forward on all subsequent reports). If all cumulative reclassifications across Data Safety and Monitoring Board reports were totalled, one would arrive at an inflated total of 288 events representing the 38 independent reclassifications. If one divides this cumulative number of events (288) by the true total number of events we identified (396), one arrives near the 75% estimate noted by the panel (288/396 = 72.7%). If our proposed explanation is correct, the panel’s calculation is completely erroneous in using cumulative totals instead of the unique misclassification numbers. Only 38 patients were reclassified, as noted above, and in a nondifferential manner.
Exclusion of patients after randomization
Our original publication6 provided a detailed and transparent rationale and accounting for all 2468 patients who underwent randomization and were either included in or excluded from the primary analysis.21 Of the 137 patients who were excluded, 50 were removed because they withdrew consent before surgery, 22 patients’ surgeries were cancelled, 7 patients were excluded because the study drug was not available, and 21 patients were effectively removed from the study by their attending clinicians because they did not receive the study drug (withdrawal at the time of surgery). All withdrawals by the clinical teams were done without knowledge of drug allocation. The remaining 37 patients were removed for eligibility reasons, such as preoperative coagulation concerns identified after randomization or a change in the type of procedure just before surgery rendering them ineligible (e.g., valve repair instead of replacement). All 137 instances were deemed by the investigators to be acceptable postrandomization exclusions.21
Could the 137 exclusions have resulted in biased estimates of treatment effects? They were not related to biased treatment withdrawals — neither attending surgeons nor anesthesiologists were able to guess allocated intervention at a rate better than chance (reported κ score 0.23).22 In addition, the BART employed central randomization, blinded allocation concealment and triple-dummy blinding of study interventions to minimize bias.
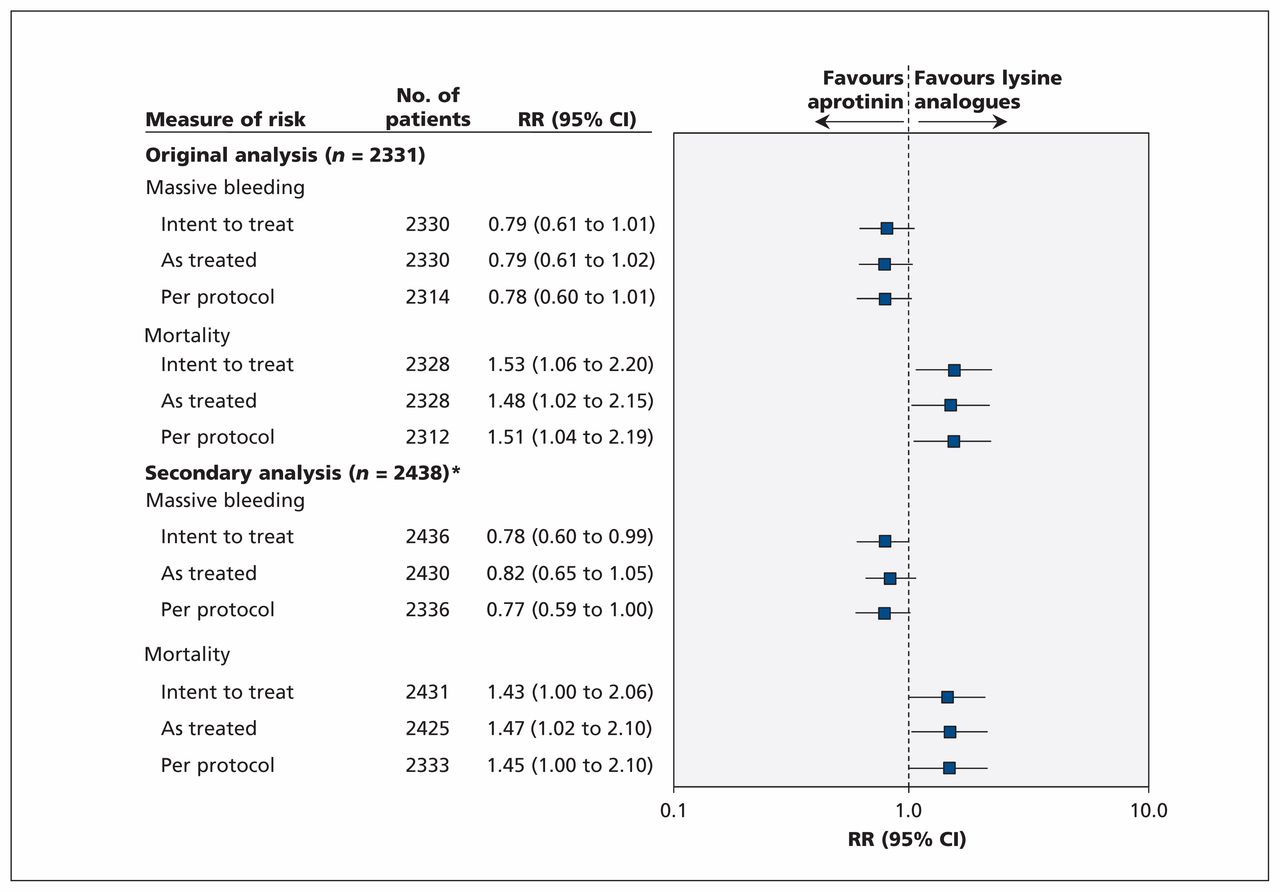
To appreciate how withdrawals could affect our conclusions, we undertook “as treated” and “per protocol” analyses. These additional analyses were also requested by the Health Canada’s expert panel. When we compared the 2438 patients (excluding only those patients for whom outcome assessment was not possible because of withdrawal of consent [n = 22] or loss to follow-up [n = 8]) according to treatment received regardless of allocation, the relative increase in death was 1.47 (95% CI 1.02 to 2.10) for aprotinin compared with both other drugs (Figure 1), compared with 1.48 (95% CI 1.02 to 2.15) from our original analysis involving 2331 patients. In addition, we documented a relative increase in death of 1.45 (95% CI 1.00 to 2.10) for all patients who underwent randomization and were given their respective allocated treatments as per protocol (n = 2333), compared with 1.51 (95% CI 1.04 to 2.19) in our original analysis (Figure 1). Clearly, there were no important qualitative or quantitative differences in effect estimates.
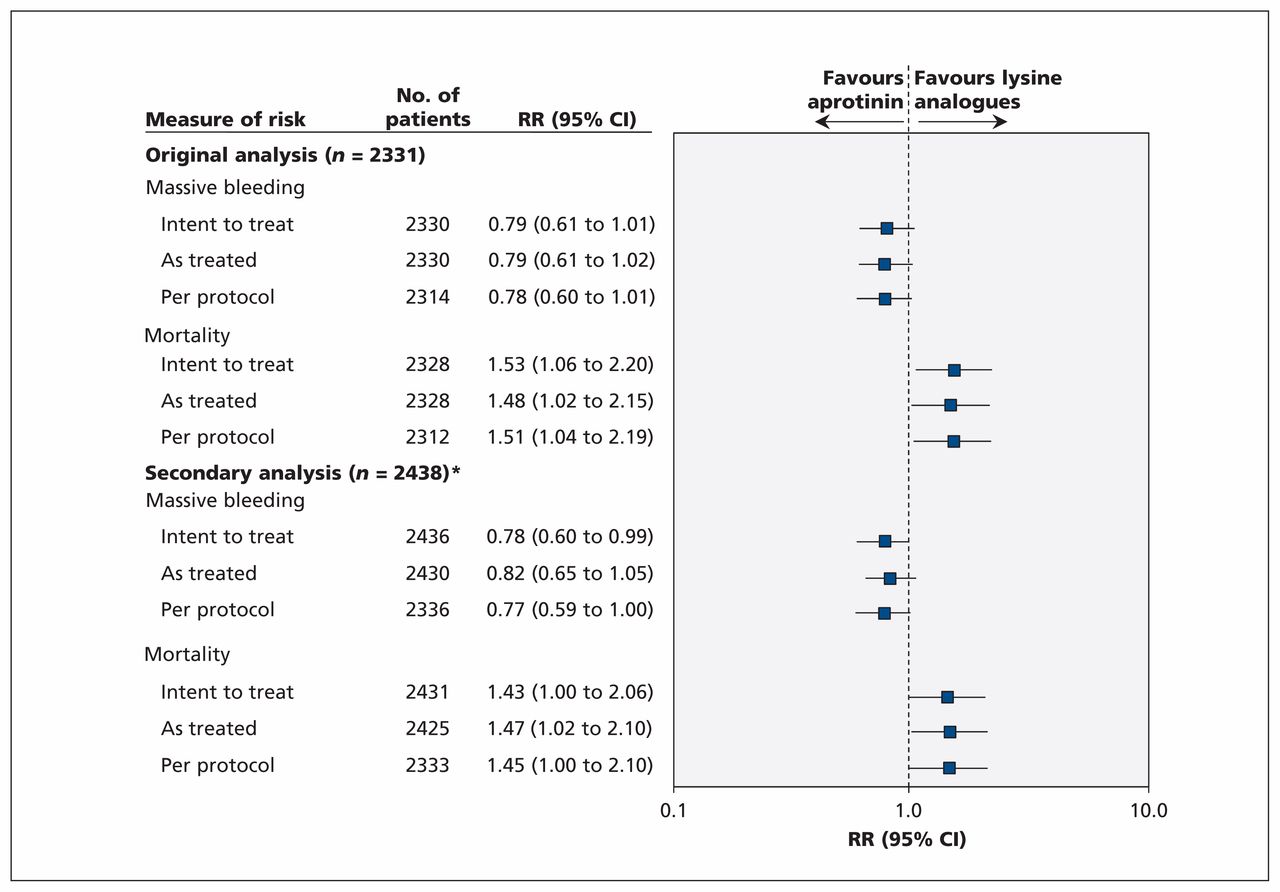
Sensitivity analyses comparing aprotinin with lysine analogues in all patients who underwent randomization in the BART. CI = confidence interval, RR = risk ratio. *Excluded only those patients for whom outcome assessment was not possible because of withdrawal of consent or loss to follow-up.
We also noted that the beneficial effects of aprotinin on massive bleeding consistently favoured aprotinin in both the as treated and per protocol analyses. Moreover, mortality (Figure 2A) and massive bleeding (Figure 2B) point estimates did not change in size or direction throughout the trial. Given the consistency of results over time, and our sensitivity analyses, the impact of the 137 exclusions on our interpretation of the results is negligible.

(A) Cumulative comparison of 30-day mortality (aprotinin v. lysine analogues). Data presented summarize the risk ratio comparing aprotinin with the lysine analogues as additional patients are incorporated into the analysis over time. (B) Cumulative comparison of massive bleeding (aprotinin v. lysine analogues). Data presented summarize the risk ratio comparing aprotinin with the lysine analogues as additional patients are incorporated into the analysis over time. CI = confidence interval, RR = risk ratio.
Baseline imbalances in heparin use and monitoring of anticoagulation
As part of its evaluation of aprotinin, Health Canada undertook a large number of secondary analyses in an attempt to explain the mortality differences we saw. In our primary report, we were unable to identify definitive reasons why aprotinin may have increased mortality, because a review of all deaths by the BART adjudication committee and exploratory analyses did not provide a clear mechanism or explanation. Faced with this same issue, Health Canada’s statistical analysis initially focused on a risk of surgery score and baseline characteristics. Health Canada statisticians elected to focus on a five-day window to assess mortality, eliminating more than a third of all events and censoring differentially between groups. They also opted to attempt to examine the effects of severity of illness and surgery using the Toronto Risk Score (for which contributing information was not purposively collected in the BART) rather than adjusting risk estimates using other accepted variables such as age, coexisting illnesses, left ventricular dysfunction and type of procedure. In doing so, Health Canada’s statisticians removed 191 patients (8.2%) from the analysis, as necessary data were not available for the modelling exercise, which may have resulted in potential biases and losses of important information. Nonetheless, using this approach, Health Canada was not able to detect important influences on mortality from more than 50 baseline characteristics examined, including severity of preoperative risk. In our primary publication, using multivariable regression techniques that included all patients, we derived similar inferences from our many analyses (Table 1, Figure 1). There were no appreciable quantitative or qualitative differences in effect size estimates for either our primary outcome or mortality when evaluating the effect of age, coexisting illnesses or surgical urgency while adjusting for sex, procedure, cross-clamp time and American Society of Anesthesiologists score (Table 1).
Assessment of risk comparisons between aprotinin and lysine analogues
Unable to explain the association of aprotinin with increased mortality, Health Canada examined associations between hemostatic variables and treatments. Differences were noted in postoperative partial thromboplastin time and international normalized ratio values, which were generally higher in the aprotinin group. Similarly, Health Canada reported that mean activated clotting times measured during surgery were prolonged with aprotinin compared with the lysine analogues. However, these values were inconsistently recorded on case report forms (values were missing from 15% to 22% of forms). In the planning of the BART, a decision had been made not to emphasize these variables because a number of reports had noted that aprotinin artificially increased partial thromboplastin times.23,24 During cardiopulmonary bypass, the practice in Canada was to monitor Kaolin activated clotting times every 30 minutes, maintain them at greater than 480 seconds throughout the bypass period and record the values on the perfusion chart. To the best of our knowledge, perfusionists followed institutional protocols assiduously. In the only values recorded in the BART database following heparinization, mean activated clotting times were 680 (standard deviation [SD] 216.9) seconds in the aprotinin group, versus 653.1 (SD 208) seconds using other antifibrinolytic agents (mean difference 27.3 s [95% CI 9.02 to 45.6], p < 0.01). Canadian clinical practice, as well as our study data, argues that inadequate anticoagulation in any group in the BART is highly unlikely.
However, the more important question was not whether monitoring was adequately reported in the case report forms, but whether heparin administration was sufficient to prevent thrombotic events leading to deaths. Health Canada’s analyses showed substantially lower mean heparin doses in patients receiving aprotinin as compared with other agents.19 However, the analysis did not adjust for weight and pump time, nor did it explore associations between treatment groups, heparin dosing and outcomes.
We performed a new series of exploratory post hoc analyses in response to the panel’s concerns about the adequacy of anticoagulation during routine surgery in Canadian centres participating in the BART. We found that overall mean heparin doses adjusted for weight and pump time were comparable between patients in each group (aprotinin group: 296.1 [SD 319.8] IU/kg per hour; lysine analogues: 294.8 [SD: 306.9] IU/kg per hour; p = 0.97). Similarly, among patients who died, heparin doses were not significantly different (mean difference 14.0 [95% CI −38.1 to 66.1] IU/kg per hour). More recent analyses done in response to these concerns, in which we investigated associations between total heparin dosages (divided into quartiles) and mortality, (Figure 3) did not yield any meaningful or consistent trends that were modified by heparin dose.

{kind=link}
{kind=link}
{kind=link}
Comparison of 30-day mortality (aprotinin v. lysine analogues), stratified by heparin dose. The adjusted analyses were done using multivariable logistic regression and accounted for the effects of age, sex, surgical procedure, total bypass duration and cross-clamp duration. CI = confidence interval, OR = odds ratio.
Conclusion
Health Canada has made inaccurate and unclear assumptions about how the BART investigators reported the trial’s interim and final results. Multiple post hoc analyses undertaken by Health Canada statisticians have further reenforced unjustified conclusions that have resulted in the reapproval of aprotinin for use in cardiac surgery in Canada. The European Medicines Agency has made limited attempts to independently substantiate or verify information provided by Canadian regulators, and BART investigators were never contacted by the agency’s evaluators. A discussion with the principal investigators and perhaps additional explanatory analyses would have ensured that any regulatory decision was based on accurate and complete information about the collection of data and planning of analyses.
To get appropriate estimates of effect in practice, pragmatic trials opt for a large sample size, limited data collection, usual care comparisons and environments, fewer exclusions and limited audit procedures. The usual care conditions adopted in the BART should not be confused with flawed study design or inadequate monitoring as suggested by the European Medicines Agency. Studies undertaken in usual care settings should be applauded as generating more useful evidence than small, tightly controlled, proof of concept studies needed for drug approval. Before the BART, all available evidence clearly documented that aprotinin decreased bleeding rates and possibly improved clinically meaningful outcomes when compared with nothing. The BART showed that aprotinin use in cardiac surgery may do more harm than good when compared with other agents, which is a far more useful comparison mimicking real-world conditions. Yet it would seem that incorporating evidence from pragmatic trials such as the BART into regulatory reviews is challenging. The BART forced regulatory authorities to either include information from all studies comparing aprotinin to active agents6,25 or base approval exclusively on placebo-controlled trials. The significant increase in the risk of death6,26,27 in comparing one active agent with another, as reflective of actual practice, was all but dismissed. Moreover, the increased risk of death noted in the primary analysis of the BART data was consistent over time (Figure 2A), which suggests a robust estimate.
We consider that the prudent regulatory response to uncertainty would have been to mandate a second large trial comparing aprotinin to an active agent to either refute or confirm results from the BART.
KEY POINTSAfter aprotinin’s initial removal from the market in 2007, Bayer was again allowed to market the drug in Canada and Europe in 2011.
This decision was based on criticisms of the Blood Conservation Using Antifibrinolytics in a Randomized Trial (BART), which the trial’s authors refute.
The authors of the BART suggest that the prudent regulatory response to any uncertainty would be to mandate a second large trial comparing aprotinin to an active agent.
Footnotes
Competing interests: Brian Hutton has received honoraria from Amgen Canada. C. David Mazer has received honoraria and research support from Bayer, Cubist, The Medicines Company, NovoNordisk, AstraZeneca, Bristol-Myers Squibb, Oxygen Biotherapeutics, Hemocon, Quark Pharmaceuticals, Hospira and Fresenius-Kabi. No other competing interests were declared.
Disclaimer: Morris Blajchman and George Wells are members of CMAJ’s Editorial Board and were not involved in the editorial decision-making process for this article.
This article has been peer reviewed.
Contributors: Paul Hébert, Dean Fergusson, Brian Hutton, David Mazer, Stephen Fremes, Morris Blajchman, Charles MacAdams, George Wells, Jim Robblee and Kevin Teoh contributed to the conception and design of the study. Paul Hébert, Dean Fergusson, Brian Hutton, David Mazer, Stephen Fremes, Charles MacAdams, Jim Robblee, Jean Bussières and Kevin Teoh acquired the data. Paul Hébert, Dean Fergusson, David Mazer, Stephen Fremes, Morris Blajchman, Charles MacAdams, George Wells, Jim Robblee, Jean Bussières and Kevin Teoh analyzed and interpreted the data. Paul Hébert and Dean Fergusson drafted the manuscript and revised it critically for important intellectual content. All of the authors reviewed the manuscript, approved the final version submitted for publication and have agreed to act as guarantor of the work.
Data sharing: Readers may request data from the authors. The authors agree to share data underlying the reported analyses on reasonable request. This would be carried out in public in correspondence of cmaj.ca.