


A 26-year-old black woman underwent evaluation in the emergency department after two weeks of worsening nausea, vomiting and fatigue. Her medical history included having had malaria at eight years of age, for which she had received treatment in Eritrea. A complete review of systems was unremarkable aside from a two-day history of a mild bitemporal headache. The patient stated no recent changes in her dietary habits or bowel movements. She did not take any medication or over-the-counter products, and she had never used illicit drugs. She had no history of venous thromboembolism.
Our patient’s vital signs were as follows: temperature 36.7°C, heart rate 87 beats/min, blood pressure 105/71 mm Hg, respiratory rate 16 breaths/min and oxygen saturation 100% on room air. She appeared fatigued and showed signs of pallor on her palms and nail beds. A complete neurologic examination that included cranial nerves, gait, reflexes, proprioception and sensation was unremarkable. The rest of the physical examination results were normal, including the absence of hepatosplenomegaly.
Laboratory investigations in the emergency department showed normocytic anemia (mean corpuscular volume [MCV] 89.9 [normal 80–96] fL), with hemoglobin 57 [normal 120–152] g/L, mild thrombocytopenia (platelets 119 [normal 150–400] × 109/L) and normal leukocyte count and differential. A preliminary workup for anemia showed elevated lactate dehydrogenase (> 1800 [normal 110–220] U/L) and mildly elevated total bilirubin (23 [normal 3–17] μmol/L), with a conjugated bilirubin of 6 (normal 0–5) μmol/L. The reticulocyte count was normal (26 [normal 7–31] × 103/L). These results, taken in totality, were consistent with hemolytic anemia. There were no previous laboratory results for comparison.
What further evaluation should this patient undergo?
Fibrinogen level
Coomb test
Peripheral blood smear
Malaria antigen test
All of the above
The answer is (e). All of the above are appropriate diagnostic tests for further workup of a hemolytic anemia for this patient. We excluded disseminated intravascular coagulation on the basis of a normal fibrinogen level and normal coagulation studies. To evaluate for autoimmune hemolytic anemia, we ordered a direct Coomb test, which was negative. This result, combined with a normal total complement test, allowed us to conclude that a diagnosis of autoimmune hemolytic anemia was unlikely. Direct Coomb tests have a sensitivity of 90%–95% and specificity of 99% in diagnosing autoimmune hemolytic anemia.1 We chose to order a rapid malaria antigen test even in the absence of recent travel, given our patient’s medical history. The test was negative. To assess for underlying microangiopathic hemolytic anemia, we requested a peripheral blood smear, which showed polychromatophilia, macro-ovalocytes, hypersegmented neutrophils and large platelets (Figures 1 and 2). In addition, schistocytes and fragmented cells were present (Figure 2).
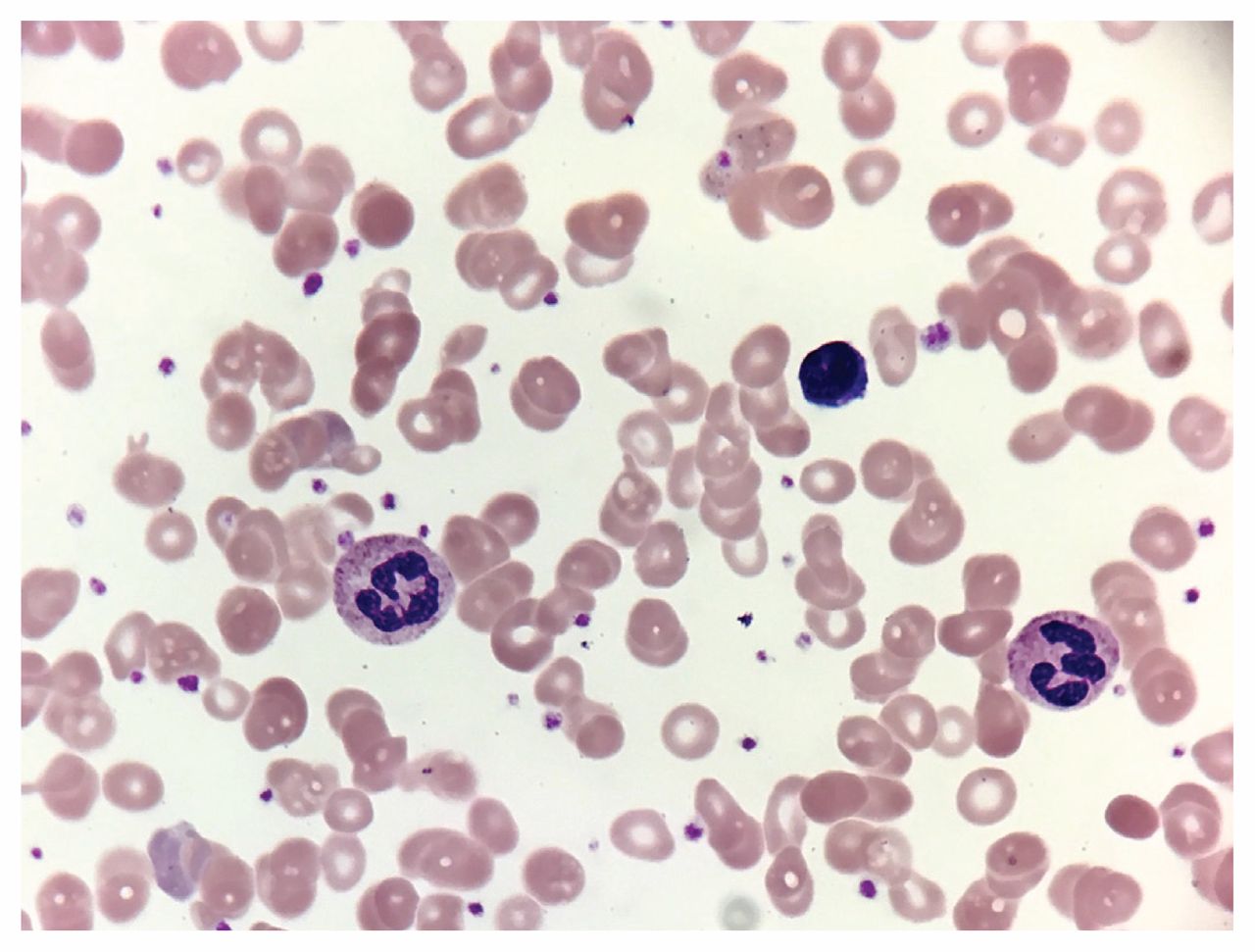
Peripheral blood smear from a 26-year-old black woman showing anisocytosis, poikilocytosis, macro-ovalocytes, hypersegmented neutrophils and large platelets.
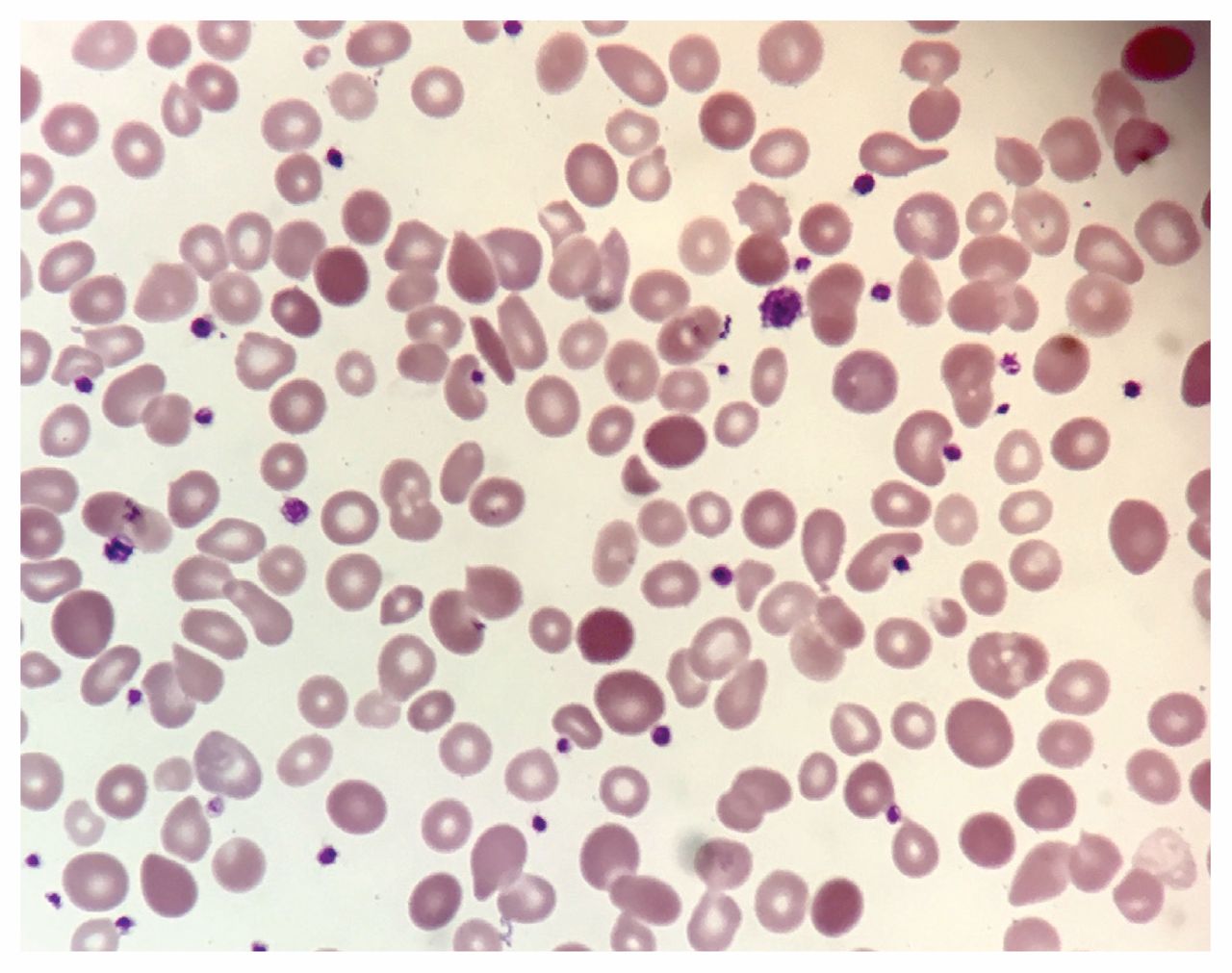
Peripheral blood smear from the same patient showing macro-ovalocytes and fragmented cells.
Given the results of the peripheral blood smear, which of the following diagnoses should be considered?
Microangiopathic anemia
Sickle cell crisis
Thalassemia
Paroxysmal nocturnal hemoglobinuria
Cobalamin deficiency
The answer is both (a) and (e). Both microangiopathic anemia and cobalamin (vitamin B12) deficiency should be considered. Although our patient was originally from Sub-Saharan Africa, she had no history of sickle cell crisis and, with an absence of sickled red blood cells on peripheral smear, this diagnosis remained low on our differential. Paroxysmal nocturnal hemoglobinuria was possible given the presence of hemolysis with iron deficiency; however, it was unlikely, given our patient’s negative history for venous and arterial thromboembolism, and the absence of leukopenia. Thalassemia could present with hemolytic anemia; however, our patient’s peripheral smear showed macrocytosis as opposed to microcytic hypochromic anemia, which is classically seen in thalassemia.
The presence of schistocytes and fragmented cells supported a diagnosis of microangiopathic hemolytic anemia. At this point, our differential diagnosis included thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (HUS) given the presence of thrombocytopenia and the findings consistent with microangiopathic hemolytic anemia on the peripheral blood smear. Thrombotic thrombocytopenic purpura was lower on our differential given the absence of other associated features, such as altered mental status, renal failure and fever. However, our patient presented with a bitemporal headache and mild thrombocytopenia, which cautioned us to keep this life-threatening diagnosis in mind. Regarding HUS, the patient did not have any history of food-borne illness, exposure to a contaminated water supply or any preceding episode of diarrhea. Furthermore, she had relatively mild thrombocytopenia without the evidence of acute kidney injury commonly associated with HUS. Box 1 summarizes the differential diagnosis of thrombotic microangiopathy.2
| Diagnosis | Cause | Clinical presentation |
|---|---|---|
| Acquired disorders | ||
| Thrombotic thrombocytopenic purpura | Autoantibody inhibition of ADAMTS13 activity | Diverse, including weakness, gastrointestinal symptoms, purpura and neurologic symptoms; acute kidney injury is uncommon |
| Hemolytic uremic syndrome (Shiga toxin–mediated) | Enteric infection with Shiga toxin–secreting strain of Escherichia coli or Shigella dysenteriae | More common in children; presents with acute kidney injury after an episode of diarrhea |
| Complement-mediated | Autoantibody inhibition of complement factor H activity | Acute kidney injury |
| Drug-mediated (immune reaction) | Quinine and possibly other drugs with formation of drug-dependent antibodies | Sudden onset of severe systemic symptoms and anuric renal failure |
| Drug-mediated (non-immune reaction) | Multiple mechanisms | Gradual onset of renal failure over weeks or months |
| Hereditary disorders | ||
| Thrombotic thrombocytopenic purpura | ADAMTS13 deficiency (homozygous or compound heterozygous for ADAMST13 mutations) | Usually presents in children (also possible in adults); acute kidney injury is uncommon |
| Complement-mediated | Mutations in complement genes causing uncontrolled activation of alternative complement pathway | Usually presents in children (also possible in adults); acute kidney injury is common |
| Metabolism-mediated | Homozygous mutations in MMACHC | Usually presents in children younger than 1 year; 1 case reported in an adult with hypertension and acute kidney injury |
| Coagulation-mediated | Homozygous mutations in DGKE | Usually presents in children younger than 1 year with acute kidney injury |
* Adapted from George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014;371:654–66.
We considered vitamin B12 deficiency in our differential diagnosis given the presence of megaloblastic changes on the peripheral blood smear, including macro-ovalocytes, giant platelets and hypersegmented neutrophils. Furthermore, the inappropriately normal reticulocyte count in the context of severe anemia pointed to underlying bone marrow failure. Vitamin B12 deficiency was subsequently confirmed with a serum level of 62 (normal 140–700) pmol/L. In addition, the patient was found to have concomitant iron deficiency, with a ferritin level of 38 (normal 50–300) μg/L, which could have been the reason for our patient’s normal MCV.
Given the finding of severe vitamin B12 deficiency, we gave our patient vitamin B12 parenterally and folic acid supplementation. We administered 1 unit of packed red blood cells, and her hemoglobin increased to 73 g/L with marked improvement in her symptoms. She was subsequently discharged home with vitamin B12 (1000 μg subcutaneously daily for six weeks, then monthly), folic acid (5 mg orally daily) and iron sulfate (300 mg orally daily).
At six weeks’ follow-up, her symptoms had completely resolved, with normalization of her blood counts (hemoglobin 137 g/L, platelets 279 × 109/L) and no signs of hemolysis (normal lactate dehydrogenase and total bilirubin). Of note, her serum vitamin B12 level normalized, suggesting recovery of her tissue stores.
In the context of the patient’s vitamin B12 deficiency, what additional workup should be done?
Bone marrow aspiration
Hemoglobin electrophoresis
Anti-intrinisc/antiparietal cell antibodies
Genetic testing
Methylmalonic acid and homocysteine levels
The answer is (c), anti-intrinsic/antiparietal cell antibodies. Homocysteine and methylmalonic acid are metabolic intermediates that accumulate in vitamin B12 deficiency, and are typically reserved for cases in which the initial vitamin B12 levels are borderline or inconclusive. Genetic testing should not be performed in this patient, as there is no suspicion for an underlying hereditary hemoglobinopathy. Furthermore, a bone marrow aspirate or biopsy is not appropriate to evaluate vitamin B12 deficiency, as the findings typically do not distinguish these deficiencies from other hematologic disorders. Hemoglobin electrophoresis would be appropriate for a suspicion of sickle cell anemia (b) or thalassemia.
In the context of combined vitamin B12 and iron deficiency, we strongly suspected a gastrointestinal malabsorption disorder. Our patient had no anti-intrinsic factor antibodies, but she did have antiparietal cell antibodies, which suggests but is not diagnostic of pernicious anemia. As part of the workup for her severe vitamin B12 deficiency, she underwent a gastroscopy with biopsies that were consistent with autoimmune atrophic gastritis.
Discussion
Clinical manifestations of vitamin B12 deficiency are often subtle; however, the consequences of this condition can be serious, with the potential of reversible bone marrow failure and demyelinating disease of the nervous system. Existing case reports and case series have reported substantial heterogeneity in the clinical presentation of patients with hemolysis secondary to vitamin B12 deficiency.3–11 In our review of the literature, these patients most commonly presented with fatigue, asthenia, sensory neuropathy or jaundice. A small proportion of patients, however, presented asymptomatically, with evidence of hemolysis noticed incidentally on blood work (Box 2).
| Study | No. of patients | Mean or median age, yr | Mean cobalamin level, pg/mL* | Other laboratory values | Peripheral smear | Clinical presentation |
|---|---|---|---|---|---|---|
| Acharya et al.4 | 3 | Patient 1: 55 Patient 2: 58 Patient 3: 91 | Patient 1: 167 Patient 2: 100 Patient 3: 162 | Patient 1: Hg 5.0 g/dL; WBC 3100 mm3; platelets 123 000/mm3; reticulocytes 6.3%; MCV 134 fL; homocysteine 62.4 μmol/L; haptoglobin < 6 mg/dL; LDH 3152 U/L Patient 2: Hg 7.7 g/dL; WBC 3400/mm3; platelets 99 000/mm3; reticulocytes 6%; MCV 115 fL; homocysteine 88.8 μmol/L; LDH 788 U/L; serum haptoglobin < 7 mg/dL Patient 3: Hg 6.6 g/dL, MCV 146 fL; platelets 97 000/mm3; homocysteine 129.7 μmol/L; haptoglobin < 7 mg/dL | Patient 1: Schistocytes and hypersegmented neutrophils Patient 2: Macrocytosis with anisopoikilocytosis, single hyperpigmented neutrophil/ granulocyte with schistocyte Patient 3: NR | Patient 1: Lethargy, confusion, exertional dyspnea, difficulty walking, moderate lower extremity edema Patient 2: Progressively increasing fatigue, bilateral paresthesias, scleral icterus Patient 3: Increasing fatigue, exertional dyspnea, right upper quadrant pain |
| Andrès et al.5 | 201 | 67 (SD ± 6) | Pseudothrombotic microangiopathy: 50 | Mean hemoglobin 10.3 g/dL; no hematological abnormalities (56 [28%]); pancytopenia 12 (5%); pseudothrombotic microangiopathy 5 (3%); hemolytic anemia 3 (2.5%); positive anti-intrinsic factor antibodies 3 (2.5%) | Hypersegmented neutrophils and macrocytosis in two-thirds of patients | Asymptomatic: one-third of patients; mild sensory polyneuropathy or confusion: 92 patients(46%); physical weakness: 40 patients (20%); bilateral edema of the legs: 24 patients (12%); jaundice: 4 patients (2%) |
| Andrès et al.7 | 6 | 77 (range 47–89) | 52 (normal 12–74) | Hemoglobin 68 g/L; platelets 70 × 109/L; homocysteine 16.2 μmol/L; positive anti-intrinsic factor antibodies 4 patients (67%) | Megaloblastic anemia and schistocytes | Asthenia and dyspnea: 6 patients (100%); jaundice: 5 patients (83%); mild peripheral sensitive neuropathy: 3 patients (50%); impaired mental status: 1 patient (17%); peripheral infiltrated cutaneous purpura: 1 patient (17%) |
| Chhabra et al.6 | 1 | 52 | 116 | Hemoglobin 4.2 g/dL; platelets 76 × 109/L; MCV 136.8.8 fL; reticulocytes 2.48%; homocysteine > 50 μmol/L; LDH 4050 U/L; haptoglobin < 10 mg/dL; positive anti-intrinsic factor antibodies | Macrocytic dysmorphic red blood cells with slight basophilic stippling, occasional schistocytes with hypersegmented neutrophils | Fatigue and chest pain |
| Garderet et al.8 | 1 | 38 | Undetectable | Hemoglobin 4.5 g/dL; WBC 2.2 × 103/L; MCV 90 fL; platelets 5 × 103/L; haptoglobin 7 mg/dL; total bilirubin 12 mg/dL; positive anti-intrinsic factor antibodies | Hypersegmented neutrophils, anisocytosis, schizocytes, and poikilocytosis | Asthenia |
| Noël et al.9 | 7 | 72 (range 43–78) | 45 μmol/L | Hemoglobin 42 g/L; platelets 73 × 109/L; reticulocytes 13.1 × 109/L; MCV 110.6 fL; LDH 7310 IU/L; haptoglobin 0.1 g/L; reticulocytes 13.1 × 109/L | Schistocytes | Mild confusion: 2 patients (28.6%) |
| Kollipara et al.10 | 1 | 42 | 59 | Hemoglobin 6.8 g/dL; WBC 1.3 × 109/L; platelets 112 × 109/L; LDH 1155 U/L; haptoglobin < 10 mg/dL; positive antiparietal cell and anti-intrinsic factor antibodies | Normocytic, normochromic anemia with ovalocytes; tear-shaped red blood cells | Asymptomatic |
| Prueksaritanond et al.11 | 1 | 52 | < 30 | Hemoglobin 5.9 g/dL; MCV 87.1 fL; LDH 701 IU/L; platelets 161 × 109/L; WBC 4.7 × 109/L; serum haptoglobin undetectable; homocysteine 100 μmol/L; reticulocyte index 0.4% | Marked anisocytosis and poikilocytoisis; tear drop cells, elliptocytes and multiple schistocytes | Increasing generalized fatigue and weakness; jaundice |
| Yeruva et al.12 | 1 | 22 | 60 | Hemoglobin 4.8 g/dL; platelets 91 000; haptoglobin 2 mg/dL; LDH 1868 IU/L; homocysteine 22.4 μmol/L; positive anti-intrinsic factor; positive antiparietal cell antibodies | Marked anisocytosis, hypochromasia with poikilocytosis, few schistocytes | Generalized fatigue and new onset dyspnea on exertion; pale conjunctiva with icteric sclera |
Note: LDH = lactate dehydrogenase, MCV = mean corpuscular volume, NR = not recorded, SD = standard deviation, WBC = white blood cells.
↵* Unless otherwise specified.
In the largest study involving patients with documented vitamin B12 deficiency, Andrès and colleagues examined hematologic findings in 201 consecutive patients.4 The results showed that less than half of patients presenting with vitamin B12 deficiency had the classical hematologic features of macrocytosis and hypersegmented neutrophils on peripheral blood smear. Nearly one-third of patients presented without any hematologic findings. Life-threatening hematologic findings including symptomatic pancytopenia, pseudothrombotic microangiopathy and hemolytic anemia were found in about 10% of patients.4 Other studies in the literature have reported varying hematologic findings including pancytopenia9 or autoimmune hemolytic anemia with a positive direct Coomb test result.11 Varying blood smear features have been reported, including hypersegmented neutrophils and macrocytosis, schistocytosis, normocytic and normochromic anemia, anisocytosis and poikilocytosis.3–11 In our literature review, patients who presented with hemolysis usually had vitamin B12 levels of less than 100 pg/mL. A few case reports, however, have described hemolysis in patients with vitamin B12 levels between 100 and 200 pg/mL (Box 2).
The presentation of patients with vitamin B12 deficiency can be quite diverse, with varying clinical and hematologic findings that can mimic other illnesses. The peripheral blood smear is an essential first step in investigating the cause of hemolysis, and evidence of megaloblastic changes and schistocytes should direct a physician to suspect vitamin B12 deficiency as the cause of the anemia.
Footnotes
Competing interests: Dr. Tagalakis reports personal fees from advisory boards (Sanofi, Pfizer, Bayer, Britsol–Myers Squibb and Servier), personal fees from speakers bureaus (Sanofi, Pfizer, Bristol–Myers Squibb), grants or honoraria from a commercial organization (Sanofi), and participation in a clinical trial within the past two years, outside the submitted work. No other competing interests were declared.
This article has been peer reviewed.
The authors have obtained patient consent.
Contributors: Malik Elharram and Vladimir Sapon-Cousineau contributed equally to the production of the manuscript. Vicky Tagalakis contributed to drafting the manuscript and provided editorial review of the manuscript. All of the authors reviewed the final version to be published and agreed to act as guarantors of the work.
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

