


- © 2004 Canadian Medical Association or its licensors
Abstract
ADRENOMYELONEUROPATHY IS A VARIENT OF ADRENOLEUKODYSTROPHY, both of which are rare inherited disorders of peroxisomes characterized by the accumulation of very-long-chain fatty acids in plasma, the central and peripheral nervous systems, adrenal glands and testes, which leads to dysfunction of these organs and systems. In this article, we describe an illustrative case of adrenomyeloneuropathy and discuss the clinical presentation, diagnosis and management of the 2 disorders.
Illustrative case
A 35-year-old man with a 2-year history of bilateral, progressive weakness and spasticity in his lower extremities that had been diagnosed as idiopathic spastic paraparesis was evaluated after a syncopal episode. He also reported a 2- to 3-month history of abdominal pain, occasional vomiting and poor appetite before presentation. He had erectile dysfunction and overflow incontinence requiring intermittent self-catheterization. He did not describe changes in his libido or hair growth. His past medical history was remarkable for the absence of any trauma to the spine or autoimmune diseases. He denied the use of tobacco, alcohol and illicit drugs and had not travelled outside of his province. He had no family history of neurological or autoimmune disorders.
A physical examination was significant for a supine blood pressure of 90/60 mm Hg and a pulse of 100 beats/min. The patient was afebrile. His skin was tanned, and he had pigmentation of the buccal mucosa. The respiratory, cardiovascular and abdominal examinations were unremarkable. Neurological examination revealed that the muscle bulk in his lower extremities was diminished, tone was increased and the power was reduced (3/5). Reflexes were brisk in the lower limbs, with evidence of ankle clonus. Sensation to all modalities was impaired in both lower limbs. His upper extremities appeared normal. There were no gross mental status changes or evidence of dementia, although detailed neuropsychological testing was not performed.
Laboratory investigations revealed a sodium level of 126 [normally 135–146] mmol/L and elevated levels of potassium (6.6 [normally 3.5–5.1] mmol/L), urea (19.6 [normally 3.7–7.0] mmol/L) and creatinine (150 [normally 45–125] μmol/L). The complete blood cell count was normal. Given the presence of buccal pigmentation, hypotension, hyponatremia and hyperkalemia, a diagnosis of primary adrenal insufficiency was considered; the morning cortisol level of 60 (normally 130–640) nmol/L and adrenocorticotropic hormone (ACTH) level of > 278 (normally ≤ 11) pmol/L subsequently confirmed the diagnosis. The serum testosterone level was normal.
The patient was hydrated intravenously and received glucocorticoid and mineralocorticoid replacement, and his blood pressure and electrolyte levels returned to normal. An MRI of the brain showed mild ventriculomegaly and a 1-cm area of increased signal (on T2 and FLAIR images) adjacent to the occipital horn of the right lateral ventricle and a smaller, similar lesion adjacent to the anterior horn of the left lateral ventricle. The spine was normal in appearance. Results of a lumbar puncture were normal, and nerve conduction studies were suggestive of acquired demyelinating polyneuropathy.
Of the potential causes of primary adrenal insufficiency (Table 1), adrenomyeloneuropathy (AMN) is the only one that is associated with progressive spastic paraparesis. Very-long-chain fatty acids (VLCFAs) were measured in the plasma. C26:0 levels were 2.33 (normally < 1.46) μmol/L, the C26:0/C22:0 ratio was 0.0567 (normally < 0.035) μmol/L, and the C24:0/C22:0 ratio was 1.5 (normally < 1.10) μmol/L. Given these biochemical findings and the clinical presentation, the diagnosis of AMN was established. The patient underwent genetic studies, which revealed a single sequence variation in the X-linked adrenoleukodystrophy (X-ALD) gene. At nucleotide 1587, the normal “C” nucleotide was replaced by a “T” nucleotide, which resulted in the substitution of tryptophan for arginine at amino acid 401 and was consistent with a known X-linked AMN mutation. The patient was enrolled in a program of intensive physiotherapy and rehabilitation in addition to supplemental hydrocortisone and fludrocortisone therapy; no specific treatment of the underlying AMN was initiated. Screening of the patient's family members was also recommended. On a recent 6-month follow-up visit, the patient was clinically euadrenal and had stable neurological findings.
Table 1.
Pathogenesis
AMN is a varient of adrenoleukodystrophy (ALD), both of which are rare sex-linked disorders transmitted on the X chromosome. Both conditions are characterized by progressive neurological symptoms and dysfunction of the adrenal glands and testes.1Both disorders can cause adrenal insufficiency and hypogonadism in addition to neurological symptoms. In general, ALD tends to present at an earlier age and primarily involves the brain, whereas AMN occurs later and involves the spinal cord. The frequency of X-ALD or X-AMN in the US male population is 1:42 000. The estimated combined frequency of X-ALD and X-AMN in hemizygotes and heterozygotes is 1:16 000.2
Both are disorders of the peroxisomes, which are intracellular organelles responsible for a variety of biochemical reactions, the most important being the oxidation of VLCFAs, biosynthesis of bile acids and glycoxylate detoxification.3 The oxidation of VLCFAs (hexacosanoic acid [C26:0], pentacosanoic acid [C25:0] and tetrocosanoic acid [C24:0]) in the peroxisomes involves several steps that are catalyzed by different enzymes (Fig. 1). People with ALD or AMN do not produce fatty acylcoenzyme A (acyl-CoA) synthetase, which breaks down VLCFAs in the first step in their oxidation (Fig. 1).4 VLCFAs therefore accumulate and form cytoplasmic inclusions, which leads to progressive dysfunction in the nervous system, adrenal glands and testes, all of which are sites of VLCFA metabolism. In cerebral variants of ALD, an inflammatory response is also seen, which is thought to damage myelin. Pathologically, these disorders are characterized by the presence of lamellar cytoplasmic inclusions consisting of cholesterol esterified with VLCFAs (Fig. 2).5 Siemerling and Creutzfeldt6 described the first possible case of ALD 7 decades ago, and in 1976, AMN was first reported to be a variant of ALD.7,8 Both disorders are transmitted in an X-linked fashion: the gene is located on the long arm of the X chromosome (Xq28)9 and encodes a peroxisomal membrane protein belonging to the adenosine triphosphate-binding cassette superfamily of transporter proteins. A variety of missense, nonsense mutations, single amino acid deletions, frameshifts and splice acceptor-site defects have been identified.10

Fig. 2: Electron micrograph of adrenocortical cell of a young boy with adrenoleukodystrophy, illustrating cytoplasmic inclusions of VLCFAs (arrow). Magnification х 35 000. Photo: Courtesy of Dr. James Powers, University of Rochester, Rochester, NY
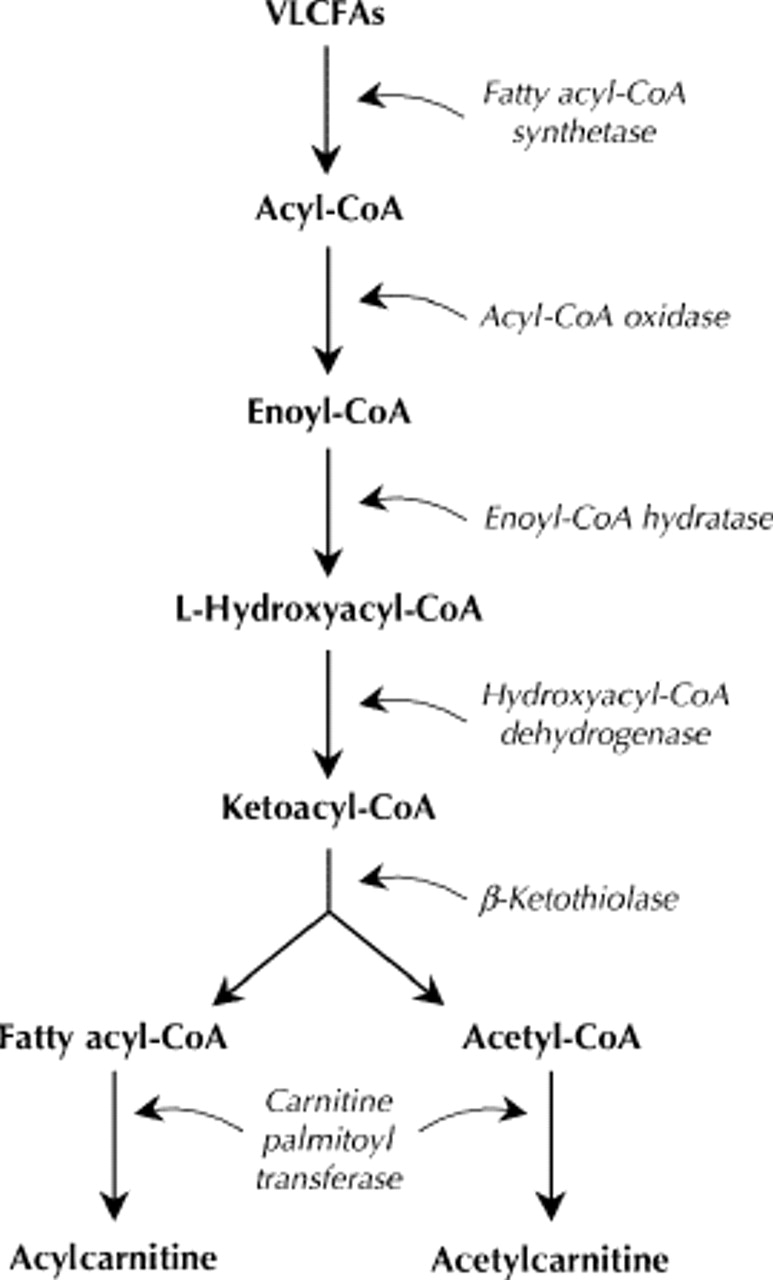
Fig. 1: The catabolism of very-long-chain fatty acids (VLCFAs) in peroxisomes. People with adrenoleukodystrophy or adrenomyeloneuropathy do not produce acylcoenzyme A (acyl-CoA) synthetase, which breaks down VLCFAs to their CoA esters in the first step in their oxidation.
Clinical presentation
There is significant clinical heterogeneity in ALD, and AMN is one of several variants (Table 2).11 The 2 most frequent phenotypes are childhood cerebral ALD and AMN, which account for about 45% and 25% of all cases respectively. Recent evidence, however, suggests that AMN may be the more common form.12 Classic ALD is characterized by rapidly progressive cerebral demyelination leading to spastic tetraparesis, dementia, seizures, and visual and auditory dysfunction. The adolescent and adult cerebral forms of ALD resemble the classic form but are later in onset. Neurological symptoms in AMN generally occur in the third and fourth decade and are characterized primarily by involvement of long ascending and descending tracts of the spinal cord and peripheral neuropathy due to demyelination, which leads to spastic paraparesis and urinary and erectile dysfunction. Although patients with AMN generally do not have clinically significant cerebral involvement, up to 50% of males with AMN have evidence of cerebral demyelination on MRI. AMN is generally considered to be a milder variant of ALD with slower progression. Rare cases of cerebellar involvement have also been described.13 Intrafamilial phenotypic variability is present, and families have been described in which some male members have ALD and others are afflicted with other clinical variants. About two-thirds of male patients have overt or subclinical adrenal insufficiency, and a significant proportion have associated gonadal dysfunction.14 As many as 50% of female heterozygotes also have neurological symptoms, and up to 80% have elevated VLCFA levels.15 Neurological symptoms in female heterozygotes usually present later, are milder than those in males and consist of mild pyramidal signs and urinary incontinence. Up to 20% of females may report severe symptoms resembling AMN. Adrenal and gonadal dysfunction is usually not present in these females, and their condition may be misdiagnosed as multiple sclerosis if a detailed family history is not taken.16
Table 2.
Diagnosis
The diagnosis of ALD and AMN is established by measurement of absolute levels of C26:0 as well as calculation of the C24:0/C22:0 and C26:0/C22:0 ratios.17 Rarely, affected males may have normal levels of C26:0, but all have abnormal ratios. There is no association between the absolute VLCFA levels and the degree of adrenal or neurological involvement. The adrenal insufficiency may either predate or follow the neurological symptoms. Measurement of cortisol and ACTH levels, ACTH stimulation test, and measurement of testosterone and gonadotropin levels may help in establishing the diagnosis of adrenal insufficiency and primary hypogonadism, respectively. An MRI may show evidence of demyelination, and abnormalities in brain stem auditory-evoked potentials, somatosensory-evoked potentials, visual-evoked potentials and motor and sensory nerve conduction velocities may be found.18 DNA linkage analysis and mutational analysis are also used. Prenatal diagnosis, with measurement of VLCFA levels in cultured amniocytes and chorion villus cells as well as DNA analysis, is also possible.19
Studies have shown that up to 35% of patients with presumed idiopathic adrenal insufficiency have elevated VLCFA levels and may later experience the associated neurological symptoms.20,21,22 These studies highlight the importance of measuring VLCFA levels in all male patients presumed to have idiopathic adrenal insufficiency; early identification may allow the opportunity for interventions to delay the subsequent onset of neurological symptoms and the opportunity for early identification of asymptomatic family members through screening. Males with “idiopathic” primary adrenal insufficiency should be evaluated for underlying ALD. In addition, males and females presenting with an unknown progressive neurological disorder involving the brain or the spinal cord should be screened for ALD. Measurement of plasma VLCFA levels is the initial screening test of choice; this is followed by genetic analysis. The presence of concomitant adrenal and gonadal dysfunction should be assessed by performing the ACTH stimulation test and by measuring the ACTH, cortisol, testosterone, luteinizing hormone and follicle-stimulating hormone levels.
Management
The management of patients with AMN is limited and includes hormonal replacement therapy for concomitant adrenal insufficiency or hypogonadism, or both. Therapeutic approaches to arrest or slow the progression of neurological dysfunction have largely been unsuccessful.23 The dietary goals are to decrease the exogenous sources of VLCFA by instituting a low-fat diet; however, dietary modifications have not been consistently shown to prevent or slow the course of the disease. Administration of monounsaturated fatty acids leads to competitive inhibition between saturated and unsaturated fatty acid precursors in the microsomal elongation pathway, thereby leading to a reduction in endogenous VLCFA synthesis.24,25 A study involving patients with ALD using the combination of dietary restriction and supplementation with glyceryl trioleate and glyceryl trierucate (Lorenzo's oil) produced significant reductions in plasma VLCFA levels but failed to convincingly change the neurological status of the patients.26 However, there was a suggestion that treatment with Lorenzo's oil may be helpful in delaying or preventing the onset in asymptomatic but genetically predisposed individuals.27 The main side effect associated with glyceryl trierucate therapy is thrombocytopenia, which occurs in up to 40% of patients. Given the lack of consistent benefit and the risks associated with therapy, it is recommended that such therapy be instituted under the aegis of a supervised research protocol. Bone marrow transplantation, in which the transplanted cells are hypothesized to provide the missing acyl-CoA synthetase enzyme, has also been evaluated as a therapeutic option. Although bone marrow transplantation has been shown to worsen the neurological status of patients with established severe disease,28it has been found to delay progression in patients with minimal disability.29 Other experimental treatments include the use of HMG–CoA (3-hydroxy-3-methylglutaryl–CoA) reductase inhibitors, cobratoxin, clofibrate, plasmapharesis, glucocorticoids, immunosuppressive agents and pentoxyfilline.30,31,32,33,34 These treatments generally have not demonstrated significant benefits.
Summary
AMN is a rare but important cause of primary adrenal insufficiency and spastic paraparesis. Although the neurological manifestations are relentlessly progressive, identification and treatment of adrenal dysfunction is life-saving. Our case highlights the importance of screening male patients with presumed idiopathic adrenal insufficiency or spastic paraparesis for underlying AMN or ALD so that the proper diagnosis can be established and genetic counselling provided for the patients and their families. With the evidence that treatment with Lorenzo's oil or bone marrow transplantation, or both, may delay progression of neurological dysfunction in patients with adrenal insufficiency and minimal neurological symptoms and in genetically predisposed family members, early screening and appropriate referral of these individuals assumes an even more important role. ALD and AMN should also be considered in female patients with a neurological disorder of unknown cause involving the brain or spinal cord.
Footnotes
-
This article has been peer reviewed.
Contributors: Stan Van Uum was involved in the initial consideration of the diagnosis of Addison's disease in this patient and in ordering tests to confirm the diagnosis; he also critically reviewed the article for intellectual content and approved the version submitted for publication. Hasnain Khandwala was the attending endocrinologist responsible for analysis and interpretation of the data leading to the the diagnosis of adrenomyeloneuropathy; he reviewed the article for intellectual content, revised the article and approved the version submitted for publication. Regina Taylor-Gjevre was the attending physician at the time of the patient's admission to hospital and assisted wth the preparation of the case report. Monika Spurek was the junior internal medicine resident on-call who was the first to assess, interview, examine and manage this patient in the emergency department; she collected the data for the case report and prepared the final version of the article.
Acknowledgements: We thank Dr. James M. Powers, Professor of Pathology and Neurology, University of Rochester, Rochester, NY, for providing us with the photomicrograph of cytoplasmic inclusions.
Competing interests: None declared.
Correspondence to: Dr. Hasnain M. Khandwala, Division of Endocrinology, Rm. 3654, Royal University Hospital, 103 Hospital Dr., Saskatoon SK S7N 0W8; fax 306 966-7926; hasnain.khandwalasaskatoonhealthregion.ca
References
In this issue
{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles

