


Abstract
Drug interactions commonly occur in patients receiving treatment with multiple medications. Most interactions remain unrecognized because drugs, in general, have a wide margin of safety or because the extent of change in drug levels is small when compared with the variation normally seen in clinical therapy. All drug interactions have a pharmacokinetic or pharmacodynamic basis and are predictable given an understanding of the pharmacology of the drugs involved. Drugs most liable to pose problems are those having concentration-dependent toxicity within, or close to, the therapeutic range; those with steep dose-response curves; those having high first-pass metabolism or those with a single, inhibitable route of elimination. Knowing which drugs possess these intrinsic characteristics, together with a knowledge of hepatic P-450 metabolism and common enzyme-inducing and enzyme-inhibiting drugs, can greatly assist physicians in predicting interactions that may be clinically relevant. This article reviews the pharmacology of drug interactions that can occur with hydroxymethylglutaryl - coenzyme A (HMG-CoA) reductase inhibitors (statins) to illustrate the scope of the problem and the ways in which physicians may manage this important therapeutic class of drugs.
Background
All important drug interactions, with the possible exception of idiosyncratic or allergic reactions, have a pharmacokinetic or pharmacodynamic basis, or both.[1, 2] Pharmacokinetic interactions refer to those where drugs or other factors cause an alteration in the concentration of unbound drug acting on the tissues. They include interactions that may lead to changes in drug absorption, drug distribution (either through binding to plasma proteins or, more importantly, binding and uptake into tissues) and drug elimination. Pharmacodynamic interactions refer to those where changes occur in tissue sensitivity or response to the same unbound drug concentrations.
The consequences of a drug interaction depend upon patient-related as well as drug-related factors (Fig. 1).3 These include the magnitude and direction of the concentration or effect changes, as well as the steepness and separation of the dose-response of the drug's intended (therapeutic) and unintended (adverse) pharmacologic actions.1 Large changes in the concentration or tissue response to a drug possessing a flat dose-response relation or low intrinsic toxicity may be of little clinical importance. Alternatively, small changes in the concentration of potent or highly toxic drugs can be disastrous. Individual susceptibility to adverse drug effects because of health- (e.g., age, pregnancy) and disease-related factors (e.g., renal, hepatic, CNS) should also be considered. As well, the body may minimize a drug's effect through offsetting changes in tissue sensitivity, by up-regulation or down-regulation of receptor numbers or by changes in receptor-effector coupling, or both.4 What might produce minimal impairment on one occasion could be incapacitating on another occasion or in a less tolerant individual.

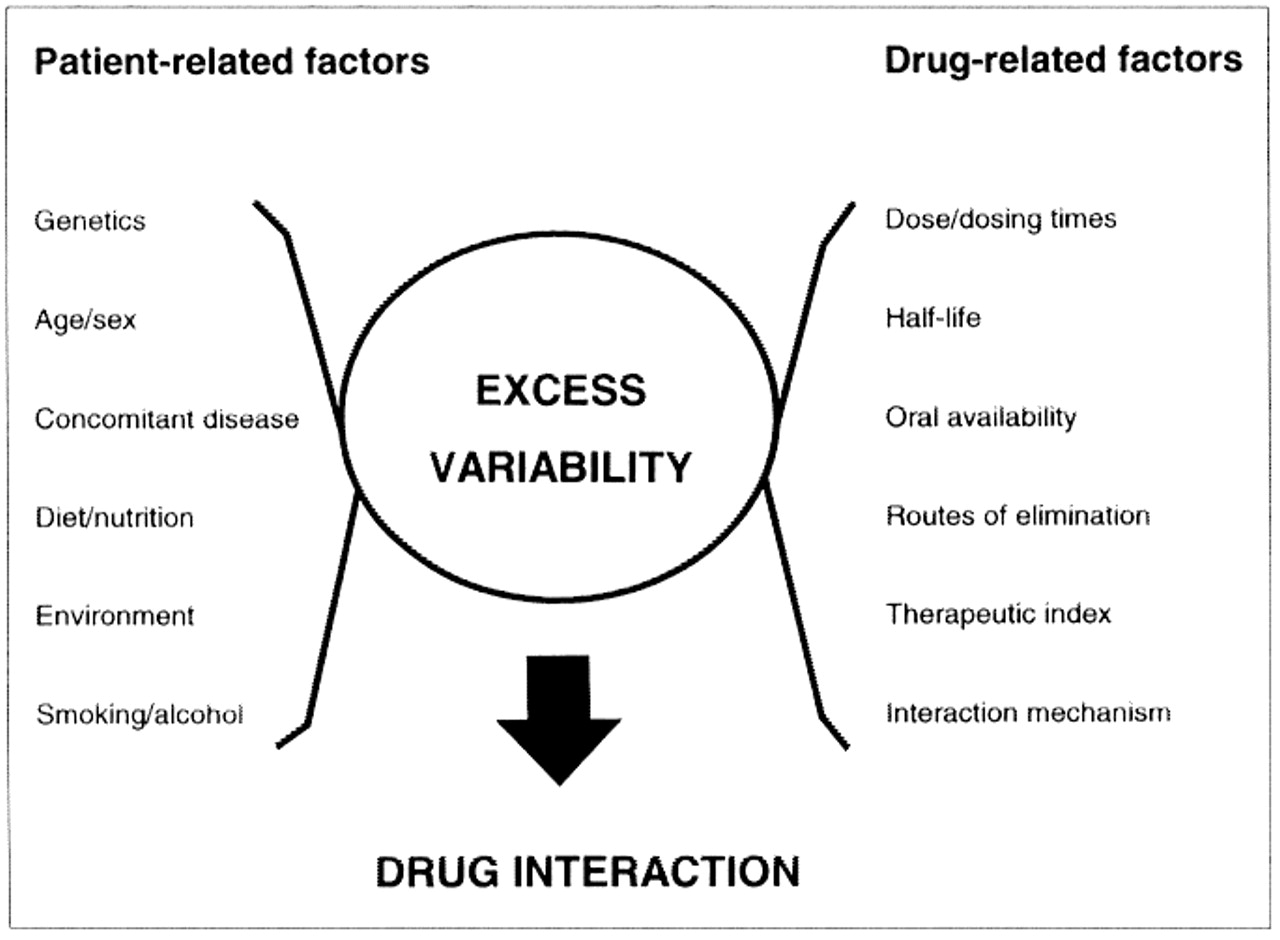{kind=link}
Fig. 1: Factors influencing drug interactions. (Adapted from Hansten).3
Interactions between drugs binding to the same sites on plasma proteins are rarely associated with changes in drug response.[1, 2] The reason for this is that most of the drug exists in the body in tissue stores, mainly in muscle and fat, not in the circulation. Thus, even large decreases in the amount of drug bound to plasma proteins is effectively buffered by a greater distribution in peripheral tissues with little or no change in unbound concentrations. The one exception occurs with drugs possessing small distribution volumes, like warfarin, where binding interactions confined largely to the circulation produce large changes in unbound concentrations and drug effects. What is important to remember is that laboratories usually report total drug concentrations and not unbound drug concentrations. Therefore, target ranges of clinically monitored drugs should be adjusted downwards in the presence of a binding interaction, normally with no change in dosage. Similar considerations apply if the levels of albumin or other binding proteins are not within the expected range.
In contrast, displacement from tissue-based binding sites or the inhibition of carrier-mediated uptake into tissues can produce large changes in unbound drug concentrations.2 Drugs and their metabolites move out of tissues as readily as they move in, and muscle and fat often contain large body stores, particularly following multiple dosing. The factors causing redistribution from tissues into the circulation are not well understood, although evidence suggests that this occurs commonly with lipophilic drugs that have large distribution volumes.5 Examples of clinically relevant interactions involving the inhibition of drug distribution and transport include the 2- to 3-fold elevations in digoxin serum concentrations following the concomitant administration of quinidine or verapamil.6
The inhibition or induction of hepatic drug metabolism is a major source of variability in drug response and is the basis of many adverse drug interactions.7 Paramount to an understanding of this is a consideration of the role of the liver in the overall elimination of the drug. Most drugs are removed from the body through multiple competing pathways of renal and hepatic excretion. If one or several of these become blocked because of disease or the action of another drug, clearance will diminish, dependent upon the relative contribution of the affected pathway(s) to the total elimination of the drug.8 If this occurs, steady-state concentrations and, correspondingly, drug or adverse effects rise. However, these in turn drive elimination through other pathways as long as they are unencumbered. Therefore, drugs that have few or minor alternative pathways are particularly prone to large concentration increases when elimination is impaired.
First-pass metabolism by the gut and liver is another important consideration. If a drug has low oral availability due to high presystemic elimination, there may be large increases in the amount of drug getting into the body if metabolism is inhibited. Where the parent drug is inactive and the pathway normally results in the formation of an active metabolite, drug response may diminish rather than increase when metabolism is inhibited.9 Conversely, response may be unchanged if both parent and metabolites are active - increases in the concentration of the parent offset by decreases in the metabolites.9
The mechanism of interaction is also an important factor; an interacting drug may not be a known inhibitor but merely a substrate for the same metabolic pathway and thereby produce only minor dose-dependent competition at the active enzyme site.10 In this case, the affinity of the substrate for the enzyme and the unbound concentration and half-life of the inhibiting drug are important determinants of the extent and time course of the interaction. Alternatively, inhibition may be noncompetitive or uncompetitive, wherein the effect is likely to be more complete and long lasting, requiring resynthesis of new enzyme before it can be overcome.10
The cytochrome P450 superfamily
Hepatic metabolism is served by a superfamily of oxygenases known as the cytochrome P450s. The purpose of these enzymes is to add a functional group to a drug, an environmental chemical or an endogenous molecule and, in so doing, increase either its polarity and excretion from the body or its interaction with similar enzymes. The most distinguishing characteristic of the cytochrome P450 family is its great diversity; members have a broad and overlapping substrate specificity and an ability to interact with almost any chemical species. The superfamily, referred to as the CYP enzymes, is subdivided according to the degree of homology in amino acid sequences. CYP enzymes possessing more than 40% homology are grouped together into families, which are designated by an Arabic numeral (e.g., the CYP1 family). Families are further divided into subfamilies, which are designated by a letter after the number (e.g., CYP2C and CYP2D subfamilies); members of each subfamily have more than 55% homology with one another. Finally, individual members are given an additional number (e.g., CYP3A4) to identify a specific enzyme pathway. Over 70 CYP families have been identified to date, of which 14 are known to occur in all mammals.11 Of the 26 mammalian subfamilies, the CYP2C, CYP2D and CYP3A subfamilies are involved in the metabolism of most clinically relevant drugs. Important substrates, inducers and inhibitors of the major CYP enzymes are listed in Table 1.
Table 1: Inducers and inhibitors of major CYP enzymes
The CYP2C subfamily comprises about 20% of all of the cytochrome P450s in the liver.12 At least 6 different CYP2C isozymes have been characterized, each having greater than 80% homology with distinct but overlapping substrate specificity. Prostaglandins and sex steroids are important endogenous substrates of the CYP2C subfamily. The most abundant enzyme in this subfamily, CYP2C9, is responsible for the breakdown of a number of drugs including ASA and many of the nonsteroidal anti-inflammatory drugs, sulfonamides, phenytoin and S-warfarin (the more active enantiomer of warfarin). CYP2C19 is involved in the metabolism of diazepam, omeprazole and the tricyclic antidepressants. Both CYP2C9 and CYP2C19 are polymorphic, meaning the expression of these enzymes is under strong genetic influence and some individuals have markedly deficient activities. Indeed, 3% of white people and 20% of all those of Japanese descent lack CYP2C19 and are unable to metabolize diazepam and omeprazole by the usual pathways.[13, 14] However, since many of the enzymes in this family have overlapping substrate specificities, it is unusual to see excessive or adverse drug effects even in people completely deficient in CYP2C19.15 Serious interactions occur predominantly with drugs that have a low therapeutic index such as warfarin or phenytoin.10
CYP2D6 accounts for only 4% of hepatic CYP enzymes,12 but is more unique in its metabolic profile. Important substrates for this enzyme include tricyclic antidepressants, selective serotonin reuptake inhibitors, neuroleptics, opioid analgesics and several of the β-adrenergic blockers. Seven to 10% of white people and 3% of black and oriental people are known to be deficient in the CYP2D6 enzyme, the so-called sparteine-debrisequine, poor metabolizer polymorph.[13, 14] These individuals show great variability in clinical response (up to 1000-fold) and commonly have adverse effects to standard doses of drugs metabolized by this enzyme. Also, they are unable to convert codeine, oxycodone and hydrocodone to their active metabolites16 and thereby derive little or no analgesic benefit from oral morphine analogues. Levels of CYP2D6 are not affected by age, sex or smoking status.17 Inhibitors are quinidine, ketoconazole and most antidepressants and neuroleptics, and there are no known inducers of this enzyme.
The CYP3A subfamily, like CYP2D6, is involved in the metabolism of a large number drugs and other chemicals and is involved in many drug-drug and drug-food interactions. It is the most abundant of all of the P450s in the human liver (25%-28%, but sometimes as high as 70%) and is widely expressed throughout the gastrointestinal tract, kidneys and lungs.12 More than 150 drugs are known substrates of CYP3A4, the major CYP3A isozyme, including many of the opiate analgesics, steroids, antiarrhythmic agents, tricyclic antidepressants, calcium-channel blockers and macrolide antibiotics. Although several substrates show age-dependent reductions in elimination, the enzyme itself does not appear to be altered.18 Also, sex-related effects are small and probably not important. Ketoconazole, itraconazole, erythromycin, clarithromycin, diltiazem, fluvoxamine, fluoxetine, nefazodone, cyclosporine and dihydroxybergamottin and various substances found in grapefruit juice, green tea and other foods are potent inhibitors of CYP3A4 and are known to be responsible for many drug interactions.[10, 15] Terfenadine, astemizole, cisapride, cyclosporine and many of the hydroxymethylglutaryl - coenzyme A (HMG-CoA) reductase inhibitors are potentially toxic drugs or drugs susceptible to large changes in concentration following enzyme inhibition and, therefore, are candidates for serious interactions with other substrates of CYP3A4.10 These interactions can have serious clinical consequences.
Interactions with HMG-CoA reductase inhibitors
The HMG-CoA reductase inhibitors (statins) are associated with 2 uncommon but important side effects, namely asymptomatic elevation in liver enzymes and skeletal muscle abnormalities, which can range from benign myalgias to myopathy (10-fold elevation in creatine kinase with muscle pain or weakness) and life-threatening rhabdomyolysis.[19, 20] The incidence of myopathy in patients taking statins alone is estimated to be 0.1%-0.2%,[20, 21] and rhabdomyolysis is exceedingly rare. Evidence suggests that myopathy is a direct consequence of HMG-CoA reductase inhibition[22, 23] and is dose-dependent.[24–27] Myopathy is most likely to occur when statins are administered with other drugs or chemicals that are themselves myotoxic or that elevate the concentrations of the statin to the toxic range. Indeed, the incidence of muscle disorders increases over 10-fold when statins are given with gemfibrozil,[20, 28–31] niacin,[20, 32] erythromycin,33 itraconazole,[34, 35] cyclosporine,[20, 36, 37] and diltiazem38 among others.
Six statins are currently marketed for the treatment of dyslipidemia in North America. Lovastatin, simvastatin, atorvastatin and cerivastatin are all substrates of CYP3A4[39–41] and would be subject to marked inhibition of metabolism by azole antifungal agents, macrolide antibiotics, selective serotonin reuptake inhibitors, cyclosporine, diltiazem and grapefruit juice. Fluvastatin is metabolized by CYP2C9; it would not be affected by these substrates, but rather would have a different spectrum of interactions,[32, 42] perhaps less clinically relevant because of the overlap between CYP2C isozymes. Pravastatin is not significantly metabolized by CYP and would be comparatively devoid of these effects.[43, 44] Lovastatin, simvastatin and atorvastatin are all extensively metabolized on first-pass through the liver[39, 40, 45] with resultant low oral availability (5%-10%), whereas cerivastatin has an intermediate availability of around 60%.46 Moreover, the active CYP3A metabolites of atorvastatin and cerivastatin contribute in large measure to their overall clinical activity.[40, 46] Thus, inhibition of firstpass metabolism of lovastatin or simvastatin could result in 10-20 fold elevations (oral availability increasing from 5% to 100%) in steady-state concentrations with a marked liability to drug toxicity. Inhibition of metabolism of atorvastatin and cerivastatin, on the other hand, is likely to produce a balanced inhibition with small changes in the total active drug concentration within the normal dosing range. Indeed, pharmacokinetic interactions of these types have been confirmed recently for each of the marketed statins.[47–53]
A MEDLINE review of all interactions involving a statin and any other drug between 1984 and 1999 revealed 1 case report of rhabdomyolysis in a patient receiving pravastatin and fenofibrate, but 27 cases of rhabdomyolysis in patients on simvastatin combined with either gemfibrozil, nefazodone, cyclosporine, itraconazole or mibefradil and 37 cases in those on lovastatin plus gemfibrozil, niacin, cyclosporine, itraconazole or erythromycin (references available on request). There are numerous other reports documenting lesser degrees of myopathy, myalgia and asymptomatic elevations in creatine kinase showing the same pattern of predilection for lovastatin and simvastatin. However, the mere potential for a drug interaction to occur, even its citation in the literature, provides little indication of the true incidence of adverse outcomes in routine clinical use. Monotherapy with lovastatin, pravastatin and simvastatin has a proven record of safety and efficacy in large clinical trials.[21, 54, 55] Moreover, there are numerous reports in the recent literature documenting the safe use of low dose statin-cyclosporine and statin-fibrate combinations in high-risk patients or patients with complex dyslipidemias[56, 57] (other references available on request). Indeed, patients who experienced serious toxicity often received other drugs, in addition to the interacting drug cited, that competed with the statin through CYP3A4.
Finally, the interaction of the statins with the fibric acid lipid-lowering agents like gemfibrozil and fenofibrate is thought to have a pharmacodynamic rather than a pharmacokinetic basis. Although rhabdomyolysis has been reported most frequently with lovastatin-fibrate combinations, there have also been cases reported with each of the other marketed statins, except possibly cerivastatin. Studies have not found any fibrate-dependent alterations in statin concentrations, however.[28, 58] Moreover, statin-induced myopathy is seen with hypothyroidism[59–61] or congenital or acquired myopathic conditions.[62, 63] This drug-disease interaction likely represents a statin-related functional mitochondrial deficit in addition to an inherent tendency toward muscular disease.
Summary
Drug interactions commonly occur in patients taking multiple medications. Although there may be some differences in the potential for statin preparations to be involved in serious adverse drug reactions, in general, they have a proven record of safety and efficacy in large clinical studies. Nonetheless, concern is warranted when statins, particularly lovastatin and simvastatin, are used in multidrug regimens because of dose-dependent toxicity and their propensity toward marked elevations in concentration if taken with drugs that inhibit first-pass metabolism.
Competing interests: Dr. Herman received speaker fees from Bristol-Myers Squibb.
Acknowledgments
A society of physicians who hold positions in the management of health resources or have an interest in the management of health resources.
-
to support and develop physicians to be successful leaders in health management
-
to support physicians in their roles as managers
-
to provide a forum for physician executives to network, learn and interact
Annual Meeting; Sheraton Centre, Toronto; Feb. 26-27, 2000
For information contact: Dr. Chris Carruthers; c/o CSPE; 3540 Paul Anka Dr.; Ottawa ON K1V 9K8; email: ccmd{at}home.com; Web site: www.cma.ca/cspe
Footnotes
-
This article has been peer reviewed.
Reprint requests to: Dr. Robert J. Herman, Department of Pharmacology, University of Saskatchewan, Health Sciences Building, 107 Wiggins Rd., Saskatoon SK S7N 5E5; fax 306 966-6220.
This work was supported by an educational grant from Bristol-Myers Squibb. Bristol-Myers Squibb had no control over the content of this manuscript.
References
In this issue
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles
Collections

