
Abstract
Aims/hypothesis
The aim of the trial was to compare the efficacy and tolerability of two types of basal-bolus therapy, using either the soluble long-acting basal insulin analogue, insulin detemir, in combination with the rapid-acting analogue, insulin aspart, or NPH insulin in combination with mealtime regular human insulin.
Methods
In this 18-week, 1:1 randomised, open-labelled, parallel trial, 595 patients with Type 1 diabetes mellitus received insulin detemir or NPH insulin in the morning and at bedtime in combination with mealtime insulin aspart or regular human insulin respectively.
Results
Glycaemic control with insulin detemir/insulin aspart was improved in comparison with NPH insulin/regular human insulin (HbA1c: 7.88% vs 8.11%; mean difference: −0.22% point [95% CI: −0.34 to −0.10]; p<0.001). Self-measured 8-point plasma glucose profiles differed between the groups (p<0.001), with lower postprandial plasma glucose levels in the insulin detemir/insulin aspart group. Within-person day-to-day variation in plasma glucose was lower with insulin detemir/insulin aspart than with NPH insulin/regular human insulin (SD: 2.88 vs 3.12 mmol/l; p<0.001). Risk of overall and nocturnal hypoglycaemia (23.00–06.00 hours) was, respectively, 21% (p=0.036) and 55% (p<0.001) lower in the insulin detemir/insulin aspart group than in the NPH insulin/regular human insulin group. Body weight (adjusted for baseline and change in HbA1c) was 1 kg lower with insulin detemir/insulin aspart than with NPH insulin/regular human insulin (p<0.001).
Conclusions/interpretation
Basal-bolus therapy using insulin detemir/insulin aspart offers a better balance of control and tolerability than with NPH insulin/regular human insulin. The low variability and more physiological action profiles generated with these insulin analogues resulted in improved glycaemic control with lower risk of hypoglycaemia and no concomitant body weight increase.
Similar content being viewed by others


Introduction
The Diabetes Control and Complications Trial and other landmark studies [1, 2, 3] have shown that tight metabolic control can reduce the incidence of, and delay the development of, late complications in patients with Type 1 diabetes mellitus. However, intensive insulin therapy is associated with increased risk of daytime and nocturnal hypoglycaemia, which has been attributed to the pharmacodynamic properties of traditional human insulin preparations [4].
Intermediate and long-acting human insulin preparations, such as NPH insulin, have pronounced insulin peaks 5 to 7 hours after injection, resulting in increased risk of nocturnal hypoglycaemia and a duration of action that is too short to maintain glycaemic control throughout the night [5]. Furthermore, the dosing precision with NPH insulin is often highly variable due to inadequate resuspension [6, 7]. Regular human insulin injected at mealtimes has a slower onset and a more prolonged action than endogenous insulin, and consequently the combination of these human insulins results in high postprandial blood glucose excursions and risk of hypoglycaemia between meals and overnight [8].
Insulin analogues have been developed to enable patients with diabetes mellitus to achieve near-normal glucose levels. First, rapid-acting insulin analogues such as insulin aspart were developed to mimic the normal mealtime insulin response more closely than injection of regular human insulin, and thereby to improve postprandial glycaemic control. This advantage, together with a reduced risk of hypoglycaemic episodes, has been demonstrated in several studies [9, 10, 11, 12]. However, the short duration of action of these analogues means that they contribute little to the between-meal basal insulin level, so patients are often obliged to increase the dose of their basal preparation. As conventional human-insulin-based basal insulins have less than ideal pharmacokinetic properties, it has been suggested that the full benefits of rapid-acting analogues would not be realised until improved basal preparations became available [12]. Recently, long-acting insulin analogues such as insulin detemir have been developed to more accurately reproduce the physiological basal insulin profile.
Insulin detemir is a long-acting insulin analogue that is soluble at neutral pH. It is an acylated derivative of human insulin [LysB29(N ε-tetradecanoyl) des(B30) human insulin], which by a combination of increased self-association and albumin binding contributes to a protracted action, providing a more reproducible absorption and a prolonged action profile [13]. In several multicentre trials in which similar bolus insulin preparations were used for therapy groups, treatment with insulin detemir resulted in predictable glycaemic control with less day-to-day variation and fewer hypoglycaemic episodes than with NPH insulin [14, 15, 16].
Thus, individually, both rapid- and long-acting analogues have been shown to have clinical advantages over their traditional human insulin counterparts. The aim of this trial, therefore, was to bring together these respective components to compare a combination of the two analogues with conventional human insulin in basal-bolus therapy for patients with Type 1 diabetes mellitus.
Subjects, materials and methods
Design
Altogether, 64 investigational sites in Europe participated in this open-labelled parallel trial, which consisted of a 6-week titration period and a 12-week maintenance period. The patients enrolled were randomised to a treatment regimen with insulin detemir or NPH insulin in the morning and at bedtime in combination with insulin aspart or regular human insulin at mealtimes respectively.
Subjects
In total, 644 patients with Type 1 diabetes mellitus were screened. Of these, 46 patients failed screening, the majority not meeting the inclusion and exclusion criteria. Inclusion criteria for entering the trial were as follows: age ≥18 years, duration of diabetes ≥12 months, BMI ≤35 kg/m2, HbA1c ≤12%, total daily insulin dose <1.4 U/kg, and current treatment with any basal-bolus insulin regimen or biphasic insulin treatment for at least 6 months. Exclusion criteria included proliferative retinopathy requiring acute treatment, impaired renal or hepatic function, severe cardiac problems, uncontrolled hypertension, recurrent major hypoglycaemia, allergy to insulin, history of drug or alcohol dependence, pregnancy, and breast-feeding. The trial was approved by the local ethics committees and health authorities, and was carried out in accordance with Good Clinical Practice guidelines (International Conference of Harmonisation) and the Declaration of Helsinki. Written informed consent was obtained from all patients before any trial-related activity.
Procedures
Enrolled patients were randomised to one of two groups receiving twice-daily basal insulin treatment (in the morning and at bedtime) of either insulin detemir (100 U/ml; Novo Nordisk, Bagsvaerd, Denmark) or NPH insulin (100 U/ml; Human isophane insulin; Novo Nordisk). In addition, to add meal-related bolus insulin to the regimens, patients randomised to the insulin detemir treatment group received insulin aspart immediately before meals (100 U/ml; NovoRapid/Novo Log; Novo Nordisk), while patients randomised to the NPH insulin group received regular human insulin 30 min before meals (100 U/ml; Actrapid; Novo Nordisk). Patients were continually advised during visits throughout the trial to inject regular human insulin 30 min before meals, and insulin aspart immediately before meals, but formal compliance was not recorded. Basal and bolus insulin were administered as subcutaneous injections in the thigh and abdomen respectively. All trial products were administered using a NovoPen 3.0 (Novo Nordisk). To reduce the risk of hypoglycaemia when patients were transferred from NPH insulin to insulin detemir, and to avoid bias with respect to optimal titration between treatments, the basal insulin (insulin detemir or NPH insulin) starting dose was 70% of the person’s previous total basal insulin dose. Patients transferring from a once-daily regimen were advised to split the basal insulin dose according to local practice or to split the dose to between 25 and 30% in the morning and 70 and 75% at bedtime. At the same time they were asked to decrease the bolus insulin dose by about 25%. Patients transferred from biphasic insulin were advised to calculate their equivalent NPH dose and start titration on 70% of this dose.
During the 6-week titration period, patients received instruction on the use of a glucose meter, and basal insulin doses were individually titrated towards predefined plasma glucose targets (pre-breakfast and pre-dinner: 5.7–7.3 mmol/l). In the following weeks, optimisation of the dose ratio between basal and bolus insulin treatment was carried out and the patients were titrated towards plasma glucose targets of 5.7 to 7.3 mmol/l fasting/preprandial and 8.5 to 10.1 mmol/l postprandial (90 min after a meal).
HbA1c was recorded at trial entry (randomisation) and after 12 and 18 weeks of treatment. Fasting, pre-lunch and pre-dinner self-measured plasma glucose levels on four normal weekdays within the last week of treatment were also recorded. Furthermore, the patients performed two 8-point plasma glucose profiles in the week before trial entry and during the last week of treatment.
All hypoglycaemic episodes and adverse events during the trial period were recorded. Hypoglycaemic episodes were classified as major (requiring assistance to treat), minor (plasma glucose measurement <3.1 mmol/l) or symptoms only (no plasma glucose measurement or plasma glucose ≥3.1 mmol/l).
Body weight was recorded at trial entry and after 18 weeks of treatment. The sites were advised to calibrate the scale every 3 months.
Analytical methods
HbA1c (reference range of assay: 4.3–6.1%; coefficient of variation: <2.0%) was measured by ion exchange HPLC (Bio-Rad Diamat; Bio-Rad Laboratories, Hercules, Calif., USA) [17]. The HbA1c measurements were analysed centrally (Laboratorium für Klinische Forschung, Raisdorf, Germany). All patients measured plasma glucose using a glucose meter (Precision Xtra; MediSense, Abbott Laboratories, Abbott Park, Ill., USA).
Statistical analyses
The sample size was determined based on a two-sided t test on a 5% significance level. Assuming an SD for HbA1c of 1.2%, a true difference in HbA1c of 0.3% (absolute) and a drop-out rate of 10%, 281 patients in each treatment group were needed to achieve a power of 80%. A routine test for baseline imbalance was not performed, as this was not statistically appropriate [18].
The primary endpoint, HbA1c level after 18 weeks of treatment, was analysed by an ANOVA model with treatment, country and baseline HbA1c (at randomisation) as fixed effects. Country was included in the model to take into account possible differences between the regions. Country was chosen instead of centre due to some centres having a relatively small number of patients. Furthermore, it was considered reasonable to assume that eating patterns and insulin administration patterns would be similar within countries. As the change in HbA1c cannot be calculated by direct subtraction of raw baseline values from model-based 18-week values, it was estimated by the same model as for the primary endpoint. This ensured consistency between the two analyses. Within-person day-to-day variation in plasma glucose (fasting, pre-lunch and pre-dinner) was compared between the two treatment groups using a mixed model. The 8-point plasma glucose profiles were analysed by repeated measures ANOVA. To estimate the relative risk of hypoglycaemia, all hypoglycaemic episodes occurring during the maintenance period (the last 12 weeks of treatment) were analysed as recurrent events by a Cox regression analysis using a gamma frailty model. Nocturnal episodes (23.00 to 06.00 hours) were analysed separately. Differences in body weight between treatment groups after 18 weeks of treatment were analysed using an ANOVA model with treatment group and country as fixed effects, and change in HbA1c from baseline to end of treatment as well as body weight at baseline as covariates. A similar analysis was carried out with change in body weight from baseline to end of treatment as the endpoint, and with body weight at baseline as an additional covariate. Adverse events were evaluated by summary statistics.
Analyses were based on the intention-to-treat analysis set, where all patients are randomised and exposed to at least one dose of either trial product. A p value of less than 0.05 was considered statistically significant. Statistical analyses were performed using SAS version 8.0 on a Unix platform (SAS Institute, Cary, N. C., USA) and S-plus for Windows version 6.03.
Results
A total of 598 patients were randomised and 595 were exposed to trial products. Of these, 298 were exposed to insulin detemir/insulin aspart and 297 were exposed to NPH/regular human insulin. In the insulin detemir/insulin aspart group 289 patients (97%) completed the trial; in the NPH/regular human insulin group 286 patients (95.3%) completed the trial. A total of 23 patients withdrew during the trial, nine patients in the insulin detemir/insulin aspart group (five owing to adverse events, two owing to non-compliance and two for other reasons), and 14 patients in the NPH/regular human insulin group (one owing to adverse events, four owing to ineffective therapy, three owing to non-compliance and six for other reasons). Baseline characteristics were similar between treatment groups except for a slightly higher HbA1c level and a slightly lower fasting plasma glucose level in the insulin detemir/insulin aspart group compared with the NPH/regular human insulin group (Table 1). All but one person were of European extraction.
Glycaemic control
Mean HbA1c was significantly lower in the insulin detemir/insulin aspart group than in the NPH/regular human insulin group after 18 weeks of treatment (7.88% vs 8.11%, p<0.001; Table 2). Change in HbA1c from baseline (at randomisation) to the end of treatment was −0.50% point in the insulin detemir/insulin aspart group compared with −0.28% point in the NPH/regular human insulin group (Fig. 1). Fasting plasma glucose tended to be lower with insulin detemir/insulin aspart than with NPH/regular human insulin after 18 weeks of treatment, but this was not statistically significant (7.58 mmol/l vs 8.10 mmol/l, p>0.05; Table 2).

Change in HbA1c and hypoglycaemic episodes with insulin detemir/insulin aspart (■) and NPH/regular human insulin (□). Data are means, or means ± SE. *p=0.036, **p<0.001
Self-measured 8-point plasma glucose profiles differed between treatment groups (p<0.001) and a smoother and more stable profile with lower postprandial plasma glucose levels was observed with insulin detemir/insulin aspart (Fig. 2). After 18 weeks of treatment, approximately 50% of the patients in both treatment groups had reached the fasting and preprandial target (5.7–7.3 mmol/l).

8-point plasma glucose profiles for insulin detemir/insulin aspart (●) and NPH insulin/regular human insulin (□). Data are means ± 2SE. BB, before breakfast; B90, 90 min after breakfast; BL, before lunch; L90, 90 min after lunch; BD, before dinner; D90, 90 min after dinner; Bed, bedtime
The day-to-day within-person variation in plasma glucose, based on self-measured plasma glucose during four normal weekdays within the last week of treatment, was significantly lower with insulin detemir/insulin aspart than with NPH/regular human insulin (Table 3).
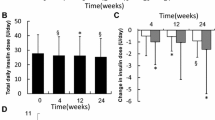
Insulin doses
After 18 weeks of treatment, the mean daily dose of basal insulin was 32.1 U (0.44 U/kg) of insulin detemir versus 28.2 U (0.38 U/kg) of NPH insulin. The mean daily dose of bolus insulin was 26.4 U (0.36 U/kg) in the insulin detemir/insulin aspart group versus 26.3 U (0.35 U/kg) in the NPH/regular human insulin group. At the end of the trial, the mean daily basal insulin dose was split between approximately 40% at breakfast and 60% at bedtime in both treatment groups.
Hypoglycaemic episodes
The occurrence of hypoglycaemic episodes per person-year was significantly lower in the insulin detemir/insulin aspart group than that in the NPH/regular human insulin group, both overall and during the night (Fig. 1). The risk of hypoglycaemia was 21% lower in the insulin detemir/insulin aspart group than that in the NPH/regular human insulin group with an estimated relative risk of 0.79 (p=0.036; Table 4). The risk of overall nocturnal and major nocturnal hypoglycaemia was, respectively, 55% lower (p<0.001; Table 4) and 83% lower (p=0.008; Table 4) in the insulin detemir/insulin aspart group than that in the NPH/regular human insulin group with estimated relative risks of 0.45 and 0.17 respectively. Similar results were achieved when data were adjusted for HbA1c (data not shown).
Body weight
A significant difference was observed in mean body weight between the two treatment groups after 18 weeks of treatment (p<0.001; Table 2). Adjusted for baseline and change in HbA1c, the mean body weight was 1 kg lower in the insulin detemir/insulin aspart group than in the NPH/regular human insulin group (p<0.001; Table 2).
Adverse events
Adverse events were equally distributed between treatments: 141 patients (47.3%) in the insulin detemir/insulin aspart group experienced at least one adverse event compared with 139 patients (46.8%) in the NPH/regular human insulin group. Serious adverse events occurred in twelve and seven patients respectively. Five patients in the insulin detemir/insulin aspart group and one person in the NPH/regular human insulin group withdrew due to adverse events. Three of the events (hyperglycaemia, allergic reaction and injection site reaction) in the insulin detemir/insulin aspart group were considered to be related to the trial product. One person treated with NPH/regular human insulin died due to a lung tumour during the trial. The event was judged unlikely to be related to the trial products.
Discussion
This trial showed that the combination of the insulin analogues, insulin detemir and insulin aspart, provides significantly better HbA1c values than NPH/regular human insulin, and that treatment with these analogues is associated with lower risk of hypoglycaemia in patients with Type 1 diabetes mellitus. In addition, administration of insulin detemir/insulin aspart resulted in more predictable plasma glucose levels with significantly lower day-to-day variation than NPH/regular human insulin and no concomitant body weight increase.
It is widely accepted that the traditional insulins used in basal-bolus therapy, NPH and regular human insulin, do not accurately reproduce the physiological insulin profile seen in non-diabetic people. For this reason, the combination of insulin detemir and insulin aspart, both exhibiting more physiological insulin profiles than NPH and regular human insulin [11, 13], was chosen to compare with the traditional basal-bolus regimen. Furthermore, both insulin aspart and insulin detemir have demonstrated certain clinical improvements over regular human insulin and NPH insulin respectively [9, 10, 11, 12, 14, 15, 16]. Therefore, it was hypothesised that the combination of these two insulin analogues in basal-bolus therapy would realise the potential of each, and demonstrate significant clinical advantages over an NPH and regular human insulin regimen. Despite possible bias to the results, an open-labelled trial design was chosen because insulin detemir, a clear solution, and NPH insulin, a cloudy suspension, are easily distinguishable from each other. This design also allowed the mealtime insulins to be administered in line with their individual recommendations. A double-blind, double-dummy technique involving twice the number of injections was considered impractical, unethical and potentially hazardous for the subjects.
The patients in both treatment groups experienced a decrease in HbA1c level during the trial with a significant difference of −0.22% point between patients treated with insulin detemir/insulin aspart and patients treated with NPH/regular human insulin after 18 weeks of treatment. Based on the DCCT study [1], Pickup et al. [19] calculated that the absolute risk reduction for sustained progression in retinopathy associated with a difference in HbA1c of 0.5% was approximately 0.5 cases per 100 patient-years. Maintaining a difference in control of 0.22% for 10 years would, according to this assumption, reduce the number of patients developing retinopathy by about 2 to 3%. Importantly, the improvement observed with treatment using insulin analogues is associated with a clear reduction in hypoglycaemic events and a small weight reduction; in contrast, optimised metabolic control in the DCCT study was associated with a considerable increase in hypoglycaemia and a weight increase [1].
Recent basal-bolus trials with rapid-acting insulin analogues and NPH have shown significant improvements in postprandial glucose control [9, 10, 20], but these improvements only translated into small improvements in HbA1c. This suggested that optimisation of the basal insulin supply by the use of basal insulin analogues could lead to further improvements in glycaemic control [21, 22]. In the current trial and earlier studies, insulin detemir has been shown to have a smooth time–action profile, meeting patients’ basal insulin requirements [13, 14]. This profile as well as the improved overall and postprandial glycaemic control obtained in this trial indicates that the combination of insulin detemir and insulin aspart provides near-normal insulin profiles, demonstrating the advantages of these new insulin analogues. Clinical trials conducted with insulin detemir have shown that clinical experience and familiarity with the time–action profile of the substances are necessary to take full advantage of the insulin analogues.
This study is the first large-scale multicentre trial to show that combination of the insulin analogues, insulin detemir and insulin aspart, in addition to a significant improvement in HbA1c, provides a lower risk of hypoglycaemia than NPH/regular human insulin treatment. The risk of hypoglycaemia is the primary obstacle to achieving tight metabolic control in intensive insulin treatment of patients with Type 1 diabetes mellitus and hypoglycaemia (especially at night). This is indeed the most feared adverse event among diabetes mellitus patients and medical staff in relation to insulin treatment [23, 24]. The DCCT documented that during intensive insulin treatment, each 10% reduction in HbA1c resulted in a 26% increase in the risk of severe hypoglycaemia, i.e. events requiring assistance [1].
In earlier trials, lower risk of hypoglycaemia with insulin detemir than with NPH has been observed with comparable HbA1c or blood glucose levels [14, 15]. This trial has shown that it is indeed possible to lower the risk of hypoglycaemia at the same time as improving glycaemic control. It is likely that the combination of rapid- and long-acting insulin analogues produces a more physiological insulin secretion and thereby reduces the risk of hypoglycaemia. The reduced risk of hypoglycaemia is also likely to be related to the more stable and predictable plasma glucose levels observed with insulin detemir/insulin aspart treatment as indicated by the lower day-to-day variability in plasma glucose. The lower fasting plasma glucose levels in the analogue group may also be expected to provide a platform that would facilitate blood glucose control for the remainder of the day, with fewer peaks and fluctuations than in the NPH/regular human insulin group.
Variability in plasma glucose levels is one of the limiting factors in determining safe plasma glucose targets with human insulins such as NPH [6, 25]. The lower within-person variability in plasma glucose levels (fasting, pre-lunch and pre-dinner) observed with insulin detemir/insulin aspart may be an important advantage, as a more predictable plasma glucose response will make titration towards more ambitious plasma glucose targets safer and more obtainable. This trial has proven that because patients can more accurately predict their glycaemic response to an injection, they can aim for tighter glycaemic targets without the worry of increasing their risk of hypoglycaemia.
The patients enrolled were titrated according to predefined plasma glucose targets (5.7–7.3 mmol/l fasting and preprandial; 8.5–10.1 mmol/l postprandial), but not all patients met the predefined targets after 18 weeks of treatment. In general, patients administered a lower proportion of the daily basal insulin dose in the morning than at bedtime. Insulin detemir has been demonstrated to have a duration of action of 20 hours at a dose of 0.4 U [13], and there seems to be scope for further increasing the basal insulin morning dose or the lunchtime prandial insulin dose to improve glucose control in the afternoon or evening (Fig. 2) to meet the later preprandial glucose targets. Recent studies with rapid-acting insulin analogues such as insulin aspart used in basal-bolus regimens suggest that these have the ability to reduce hypoglycaemia in patients with Type 1 diabetes mellitus, and that the greatest benefit is observed in patients with tight glycaemic control [22]. Owing to the lower within-person day-to-day variation in plasma glucose, further titration against the predefined targets could therefore lead to even more pronounced benefits for patients treated with insulin detemir/insulin aspart in terms of fewer hypoglycaemic episodes. On the contrary, titration towards tight plasma glucose targets with human insulins usually results in more frequent hypoglycaemic episodes [1].
As expected, the basal insulin dose in this study was higher in the analogue group due to the different bolus insulins. Prior to inclusion in the trial, around 45% of patients received one basal insulin dose in combination with mealtime rapid-acting insulin injections. This was deemed intensified insulin therapy, as no precise definition of this exists. Rapid-acting insulin analogues contribute less to the basal insulin profile, as they have a shorter duration of action than regular human insulin, and therefore the basal insulin dose must often be increased to compensate. This has been demonstrated in large-scale studies [9, 10]. Excessive weight gain has been identified as a major concern with long-term intensive therapy of Type 1 diabetes mellitus [26]. In this trial, a significantly lower mean body weight was observed in the insulin detemir/insulin aspart group than in the NPH/regular human insulin group after 18 weeks of treatment. This finding is consistent with findings from other trials with insulin detemir [14, 16]. The reason for the difference in weight development is likely to be a difference in food intake, probably due to less defensive snacking in the insulin detemir/insulin aspart group, related to the lower variability in plasma glucose and the reduced risk of hypoglycaemic episodes. Future trials may be required to focus on the potential clinical benefits and impacts of this favourable weight development with insulin detemir.
In conclusion, the combination of the insulin analogues, insulin detemir and insulin aspart, is a promising type of basal-bolus therapy in the future treatment of diabetes mellitus. The low variability and more physiological action profiles associated with these insulin analogues result in improved glycaemic control with lower risk of hypoglycaemia and no concomitant body weight increase.
References
The Diabetes Control and Complications Trial Research Group (1996) The absence of a glycaemic threshold for the development of long-term complications. Diabetes 45:1289–1298
Reichard O, Nilsson BY, Rosenqvist U (1993) The effect of long-term intensified insulin treatment in the development of microvascular complications of diabetes mellitus. N Engl J Med 329:304–309
Wang PH, Lau J, Chalmers TC (1993) Meta-analysis of effects of intensive blood-glucose control on late complications of type 1 diabetes. Lancet 341:1306–1309
The Diabetes Control and Complications Trial Research Group (1997) Hypoglycaemia in the Diabetes Control and Complications Trial. Diabetes 46:271–286
Starke A, Heinemann L, Hohlmann A, Berger M (1989) The action profiles of human NPH insulin preparations. Diabet Med 6:239–244
Kølendorf K, Bojsen J, Deckert T (1983) Clinical factors influencing the absorption of 125I-NPH insulin in diabetic patients. Horm Metab Res 15:274–278
Jehle PM, Micheler C, Jehle CR, Breitig D, Boehm BO (1999) Inadequate suspension of neutral protamine Hagedorn (NPH) insulin in pens. Lancet 354:307–335
Bolli GB, Perriello G, Fanelli CG, De Feo P (1993) Nocturnal blood glucose control in type 1 diabetes mellitus. Diabetes Care 16:71–89
Home PD, Lindholm A, Riis A (2000) Insulin aspart vs. human insulin in the management of long-term blood glucose control in Type 1 diabetes mellitus:a randomized controlled trial. Diabet Med 17:762–771
Raskin P, Guthrie RA, Leiter L, Riis A, Jovanovic L (2000) Use of insulin aspart, a fast-acting insulin analog as the mealtime insulin in the management of patients with Type 1 diabetes. Diabetes Care 23:583–588
Lindholm A, McEwen J, Riis AP (1999) Improved postprandial glycemic control with insulin aspart. Diabetes Care 22:801–805
Lindholm A (2002) New insulins in the treatment of diabetes mellitus. Best Pract Res Clin Gastroenterol 16:475–492
Pieber TR, Plank J, Goerzer E et al. (2002) Duration of action, pharmacodynamic profile and between-subject variability of insulin detemir in subjects with type 1 diabetes. Diabetologia 45 [Suppl 2]:A254 (Abstract)
Vague P, Selam J-L, Skeie S et al. (2003) Insulin detemir is associated with more predictable glycaemic control and reduced risk of hypoglycaemia than NPH insulin in patients with type 1 diabetes on a basal-bolus regimen with premeal insulin aspart. Diabetes Care 26:590–596
Hermansen K, Madsbad S, Perrild H, Kristensen A, Axelsen M (2001) Comparison of the soluble basal insulin analog insulin detemir with NPH insulin. Diabetes Care 24:296–301
Russell-Jones D, Simpson R, Bolinder J, Staedes A, Stender A (2002) Lower and more predictable fasting blood glucose and reduced risk of nocturnal hypoglycaemia with once daily insulin detemir versus NPH in subjects with type 1 diabetes. Diabetologia 45 [Suppl 2]:A51 (Abstract)
DCCT Research Group (1987) Feasibility of centralized measurements of glycated hemoglobin in the Diabetes Control and Complications Trial: a multicenter study. Clin Chem 33:2267–2271
Senn S (1994) Testing for baseline balance in clinical trials. Stat Med 13:1715–1726
Pickup J, Mattock M, Kerry S (2002) Glycaemic control with continuous subcutaneous insulin infusion compared with intensive insulin injections in patients with type 1 diabetes: meta-analysis of randomised controlled trials. BMJ 324:705
Lalli C, Ciofetta M, Del Sindaco P et al. (1999) Long-term intensive treatment of type 1 diabetes with the short-acting insulin analog lispro in variable combination with NPH insulin at mealtime. Diabetes Care 22:468–477
Madsbad S (2002) Insulin analogues: have they changed insulin treatment and improved glycaemic control? Diabetes Metab Res Rev 18:S21–S28
Heller S (2002) Reducing hypoglycaemia with insulin analogues. Int J Obes 26:S33–S36
McCrimmon RJ, Frier BM (1994) Hypoglycaemia, the most feared complication of insulin therapy. Diabetes Metab 20:503–512
Cryer PE (2002) Hypoglycaemia: The limiting factor in glycaemic management of type I and type II diabetes. Diabetologia 45:937–948
Lauritzen T, Pramming S, Gale EA, Deckert T, Binder C (1982) Absorption of isophane (NPH) insulin and its clinical implications. Br Med J 285:159–162
Purnell JQ, Hokanson JE, Marcovina SM, Steffes MW, Cleary PA, Brunzell JD (1998) Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure. JAMA 280:140–146
Acknowledgements
This trial was sponsored by Novo Nordisk, Denmark. We would like to thank the following investigators and their co-workers for their participation in the trial:
Argentina: Dr Hector Cintora, Dr Sonia Hermida
Croatia: Dr Želimir Beer
Czech Republic: Dr Milan Kvapil, Dr Jindřiška Perušičová, Dr Terezie Pelikánová, Dr Ludmila Trešlová, Dr Alena Šmahelová, Dr Alena Klimovičová, Dr Zdeněk Rušavý
Denmark: Dr Kjeld Hermansen, Dr Karsten Sølling, Dr Birger Thorsteinsson, Dr Klaus Kølendorf, Dr Erik Berg Schmidt, Dr Jens Møller, Dr Lotte Ørskov, Dr Michael Hansen-Nord, Dr Hans Perrild, Dr Henning Juhl
Estonia: Dr Hella Vides, Dr Liina Viitas
Finland: Prof. Leo Niskanen, Dr Pirkko Nyländen, Dr Toini Virkkala, Dr Liisa Hiltunen, Dr Timo Tulokas
France: Prof. Pierre Fontaine, Dr Pierre-Yves Benhamou, Dr Béatrice Bouhanick, Dr Jean-Pierre Courrèges, Dr Michel Issa Sayegh, Prof. Richard Maréchaud, Prof. Louis Monnier, Dr Helen Mosnier-Pudar, Dr Bernard Schmitt, Prof. Michel Marre
Greece: Prof. Nikolaos Katsilambros, Dr Thomas Katsaros, Dr Marian Benroubi
Italy: Dr Luigi Gentile, Dr Giuseppe Rosti, Prof. Domenico Fedele, Dr Alberto Michele Di Carlo, Dr Francesco Cannatà, Prof. Renzo Cordera
Macedonia: Prof. Gordana Pemovska
Norway: Dr Christian Fossum, Dr Thor Kristian Støle, Dr Gunnar Mouland, Dr Odd Erik Johansen, Dr Roger Apelseth, Dr Sten Reimer
Poland: Prof. Lilianna Majkowska, Dr Anna Mikołajczyk-Swatko, Dr Mirosława Polaszewska-Muszyńska, Prof. Jacek Sieradzki, Dr Ewa Jarosz-Skokowska, Prof. Roman Junik, Dr Ewa Semetkowska-Jurkiewicz
Romania: Prof. Viorel Serban
Russia: Prof. Elvira Kasatkina, Prof. Valentina Peterkova
Slovakia: Dr Milan Behuncik, Dr Zbynek Schroner
Conflict of interest for authors. All of the investigators were remunerated appropriately for their activity in this study, and in some cases for other activities undertaken together with Novo Nordisk. M.-A. Gall and G. Leth are employed by Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hermansen, K., Fontaine, P., Kukolja, K.K. et al. Insulin analogues (insulin detemir and insulin aspart) versus traditional human insulins (NPH insulin and regular human insulin) in basal-bolus therapy for patients with Type 1 diabetes. Diabetologia 47, 622–629 (2004). https://doi.org/10.1007/s00125-004-1365-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1365-z