


The rate of syphilis infection in Canada has risen in recent years.
Early neurosyphilis typically presents as meningitis or meningovascular disease, while late neurosyphilis classically causes dementia or tabes dorsalis.
Neurosyphilis may rarely mimic autoimmune encephalitis, and recognition of this is critical to ensure accurate diagnosis and prompt treatment with antimicrobial therapy.
Further study is needed to determine whether immunologic mechanisms contribute to this atypical presentation of neurosyphilis.
A 52-year-old man in a long-term, same-gender sexual relationship presented with agitation, confusion and problems speaking for about two weeks. On assessment, his vital signs were normal, but he was agitated and had global aphasia. No other focal deficits were identified on a screening neurologic examination. He suffered a witnessed generalized tonic–clonic seizure in the emergency department and was given a loading dose of phenytoin. Seizure activity stopped, but his agitation, confusion and aphasia persisted.
Six months earlier, he had been admitted to hospital with agitation, disorientation and aphasia that had developed one month after an episode of vertigo. Brain magnetic resonance imaging (MRI) had shown T2 hyperintensity of the left thalamus and medial temporal lobe (Figure 1). An electroencephalogram during this initial hospital admission showed left posterior temporal slowing, but no seizure activity. Lumbar puncture had shown inflammatory cerebrospinal fluid (CSF) with a leukocyte count of 72 × 106 cells/L with 86% lymphocytes (normal 0–5 × 106 cells/L), elevated protein of 1260 (normal 200–400) mg/L and normal glucose. His CSF bacterial culture and herpes simplex virus polymerase chain reaction were negative. Serum HIV testing was negative. Rheumatologic, paraneoplastic and autoimmune serology, including anti–N-methyl-D-aspartate receptor (NMDAR), anti–voltage-gated potassium channel, anti–glutamic acid decarboxylase (GAD), anti-Hu, anti-Yo, anti-Ri, anti-Ma2, anti-CV2 and anti-amphiphysin antibodies, was remarkable only for positive anti-GAD antibody in low titre at 2.5 (normal < 1.0) U/mL.

Magnetic resonance imaging showing left thalamic and medial temporal lobe signal abnormality in neurosyphilis in a 52-year-old man. Magnetic resonance imaging T2-weighted fluid-attenuated inversion recovery (FLAIR) shows hyperintensity of the left dorsomedial thalamus on the axial (A, arrow) and coronal (B, arrow) images. Subtle hyperintensity of the left anteromedial temporal lobe is also seen on the axial (C, arrow) and coronal (D, arrow) images.
During that admission, he had improved without any antimicrobial or immunotherapy and been discharged with outpatient speech therapy for mild word-finding difficulties. Three months later, however, he was again admitted with agitation, confusion and aphasia. A repeat electroencephalogram showed intermittent seizure activity (Figure 2) and he was started on lacosamide 200 mg and levetiracetam 500 mg, both administered orally twice daily. Electrographic seizure activity ceased, but his agitation, confusion and aphasia persisted. Repeat brain MRI at this second hospital admission showed left hippocampal atrophy without signal abnormality. Repeat serum anti-GAD antibody was again positive in low titre at 1.9 U/mL, and a repeat lumbar puncture yielded results similar to those described above. No empiric antimicrobial therapy was administered. Because of his subacute neurologic decline with seizures, medial temporal lobe signal abnormality on initial MRI, and positive serum anti-GAD antibody, a diagnosis of autoimmune encephalitis was considered. He received five days of 0.4 mg/kg per day intravenous immunoglobulin with substantial improvement, facilitating discharge home with his partner.
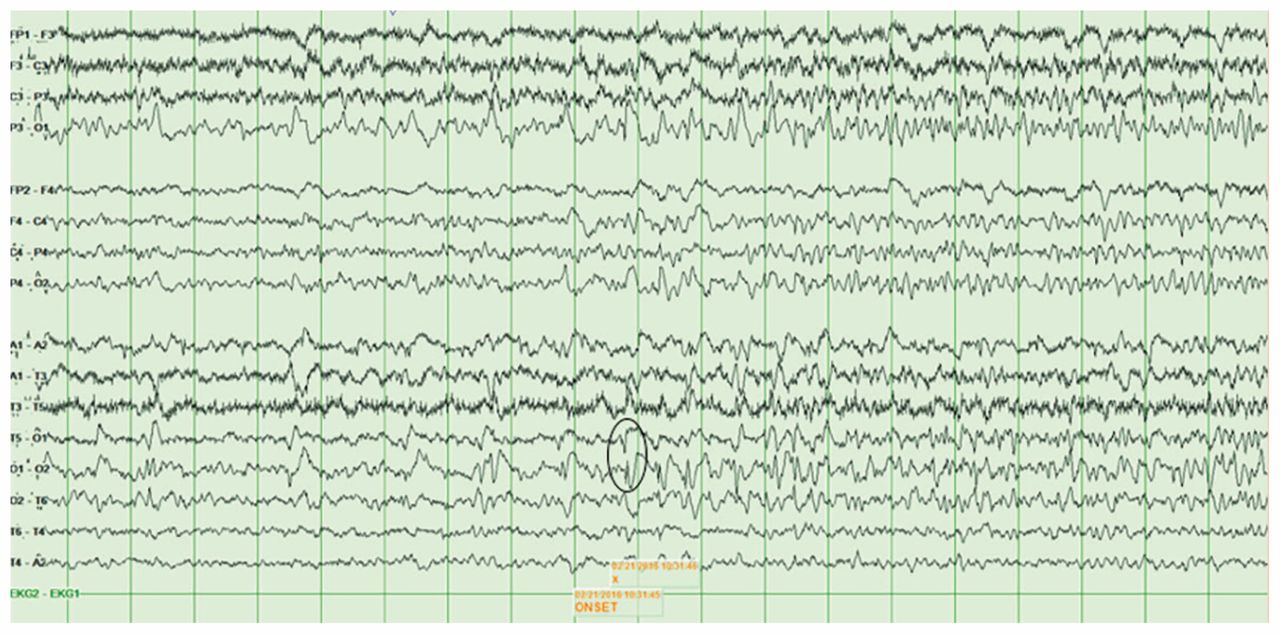
Electroencephalogram showing onset of left temporal-occipital seizure in neurosyphilis at T5-O1 and O1-O2 (circle), with propagation throughout the left temporal lobe.
Given his previous response to immunotherapy, we initiated five days of 0.4 mg/kg intravenous immunoglobulin per day, in case his symptoms were owing to autoimmune encephalitis. We also administered a seven-day course of piperacillin-tazobactam for possible aspiration pneumonia after his seizure. We noticed some improvement in his agitation and speech. However, in light of his recurrent symptoms and his relatively low anti-GAD antibody titre, we revisited the working diagnosis of autoimmune encephalitis. A repeat lumbar puncture (his third) still showed inflammatory cerebrospinal fluid, with a leukocyte count of 31 × 106 cells/L (65% lymphocytes) and elevated protein of 1669 mg/L. His CSF immunoglobulin G (IgG) index was elevated and oligoclonal bands were present, suggesting intrathecal antibody production that was compatible with autoimmune encephalitis. However, anti-GAD antibody testing in the CSF was negative.
We also wanted to rule out neurosyphilis, which remained on the differential diagnosis in the absence of serology during the previous hospital admissions. Serum treponemal automated enzyme immunoassay and serum Treponema pallidum particle agglutination assay were reactive and serum quantitative rapid plasma reagin was 128, and CSF quantitative venereal disease research laboratory test (VDRL) was 16, confirming neurosyphilis. The patient had no prior history of syphilis testing or treatment. His long-term male partner tested seronegative for syphilis. Focused history and examination of the genitals and skin showed no evidence of primary or secondary syphilis, and Argyll-Robertson pupils were not present on admission to hospital. He was treated with 4 million units of intravenous aqueous crystalline penicillin G every four hours for 30 days.
On follow-up assessment six months later, we noted sustained clinical improvement, identifying only mild neurocognitive difficulties. Repeat serum quantitative rapid plasma reagin fell fourfold, to 32. Repeat CSF testing showed a leukocyte count of only 5 × 106 cells/L (100% lymphocytes) and mildly elevated protein of 619 mg/L, and the CSF quantitative VDRL had fallen to 8, confirming response to antimicrobial therapy.
Discussion
We present an unusual case of neurosyphilis mimicking autoimmune encephalitis (Box 1),1 which has rarely been reported in the literature.2,3 Autoimmune encephalitis is the second most common cause of encephalitis with temporal lobe abnormalities after herpes simplex virus encephalitis,4 and is therefore a major diagnostic consideration in patients such as ours. Several important observations from our case may help to prevent misdiagnosis in the uncommon case of patients with neurosyphilis who present similarly, and provide insight into the pathogenesis of this unusual presentation.
Diagnostic criteria for possible autoimmune encephalitis1
Diagnosis can be made when all three of the following criteria have been met:
Subacute onset of working memory deficits, altered mental status or psychiatric symptoms
At least one of the following:
New focal central nervous system findings
Seizures not explained by previously known seizure disorder
Cerebrospinal fluid pleocytosis
MRI features suggestive of encephalitis, including T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity highly restricted to one or both medial temporal lobes
Reasonable exclusion of alternative causes, including infectious causes such as herpes simplex virus encephalitis, HHV-6 encephalitis, central nervous system Whipple’s disease, HIV encephalitis and neurosyphilis
Both our team and our patient’s previous clinicians had an initial working diagnosis of autoimmune encephalitis, in part owing to his positive anti-GAD serum antibody. However, neurologic disease related to anti-GAD antibody is associated with high serum titres (usually more than 100-fold higher than patients with type 1 diabetes mellitus) and intrathecal synthesis of anti-GAD antibody in the cerebrospinal fluid;1,5 isolated low titre positivity in serum should motivate investigation for other causes of encephalitis. An elevated IgG index and oligoclonal bands were present in the CSF of our patient, which may support a diagnosis of autoimmune encephalitis.1 Clinicians should keep in mind, however, that intrathecal antibody production may also be seen with numerous central nervous system infections, including neurosyphilis.3
Our patient responded to immunotherapy twice, including a substantial initial improvement after intravenous immunoglobulin in the absence of any antibiotic therapy. The presence of both intrathecal antibody production and response to immunotherapy in our case raises the possibility that immunologic mechanisms may have contributed to our patient’s clinical presentation. This is supported by the fact that although neurosyphilis frequently occurs in HIV-infected persons, patients with neurosyphilis mimicking autoimmune encephalitis are typically not infected with HIV.3 A competent immune system may therefore be required for this unusual presentation of neurosyphilis to occur, blurring the distinction between bacterial and immune-mediated central nervous system injury. Whether there is any role for immunotherapy in cases of neurosyphilis mimicking autoimmune encephalitis is unknown, and this question may extend to the uncommon case of patients treated with antimicrobial therapy who have neurologic worsening that is attributed to a Jarisch–Herxheimer-like reaction in the cerebrospinal fluid.2 The interface of infectious and autoimmune encephalitis has reached the forefront of neurology in recent years, as evidenced by the discovery of anti-NMDAR encephalitis triggered by herpes simplex virus encephalitis.6 Future study in this field may elucidate complex mechanisms of disease in patients such as ours.
Diagnosing and managing neurosyphilis
The rate of reported infectious syphilis cases in Canada doubled from 2.9 per 100 000 in 2003 to 5.8 per 100 000 in 2012.7 Box 2 reviews the stages of the disease.2 Neurosyphilis can occur at any time during the course of infection; early neurosyphilis typically presents as meningitis or meningovascular disease, and late neurosyphilis classically causes dementia or tabes dorsalis.2
Clinical stages of syphilis2
Early syphilis
Primary syphilis: Typically consists of painless chancre at site of inoculation, after an incubation period of about three weeks
Secondary syphilis: Refers to systemic illness with rash that classically involves the palms and soles, fever and malaise, and other symptoms, such as pharyngitis and mucous patches, weeks to months after infection
Early latent syphilis: Refers to syphilis diagnosed by serologic testing without symptoms, within one year of infection
Late syphilis
Tertiary syphilis: Refers to disease manifestations occurring 1 to more than 30 years after infection, which involve the cardiovascular system, or gummatous disease
Late latent syphilis: Refers to syphilis diagnosed by serologic testing without symptoms, more than one year after infection
Neurosyphilis (can occur during any stage of infection)
Early neurosyphilis: Occurs within months to a few years after infection, most commonly presenting as meningitis or meningovascular disease
Late neurosyphilis: Occurs years to decades after infection, typically affecting the brain (causing dementia) or spinal cord (causing tabes dorsalis)
Traditionally, non-treponemal assays (VDRL, rapid plasma reagin) have been used to screen for syphilis, followed by a treponemal assay (Treponema pallidum particle agglutination, fluorescent treponemal antibody absorption assay) to confirm infection. Recently, this approach has been reversed in Canada, with most laboratories using treponemal automated enzyme/chemiluminescent immunoassays to screen for syphilis, followed by confirmatory testing with a nontreponemal assay as well as a second treponemal assay to minimize false-positives.8 If serologic testing is positive in a patient with suspected neurosyphilis, lumbar puncture is essential to assess for CSF abnormalities that show neuroinvasive disease (leukocyte count ≥ 5 × 106/L, positive CSF-VDRL).2
Our patient’s exclusive partner of more than 30 years tested seronegative for syphilis, and he denied sexual activity outside of this relationship. It is therefore possible the patient had contracted syphilis more than 30 years ago, and would be classified as having late neurosyphilis. More rigorous contact tracing in our case was not required per the Public Health Agency of Canada guideline, because late neurosyphilis is noninfectious and is not designated a reportable disease.9
In keeping with the Public Health Agency of Canada guideline, our patient was treated with high-dose intravenous aqueous crystalline penicillin G for neurosyphilis.9 Success of antimicrobial therapy is marked by clinical improvement or stabilization, decline in CSF leukocyte count at six months and resolution of all CSF abnormalities at two years.2 Our patient had sustained clinical improvement after treatment, with both serologic and CSF analysis confirming response to therapy. Unfortunately, some neurocognitive impairment persisted, possibly due to permanent neuronal injury, shown by atrophy of medial temporal lobe structures on MRI.
In light of our experience, we suggest considering syphilis testing in patients with possible autoimmune encephalitis, especially those with temporal lobe abnormalities on neuroimaging, to avoid diagnostic delay in atypical cases such as ours. The US Centers for Disease Control and Prevention also recommends that HIV-infected persons and sexually active men who have sex with men be screened for syphilis at least annually.10
Acknowledgement
We thank Dr. Richard Chan for reviewing the manuscript.
Footnotes
Competing interests: Jorge G. Burneo reports research funding and educational grants from UCB Canada, Ontario Brain Institute and Epilepsy Ontario outside the submitted work, and was on the advisory boards of Sunovion and UCB Canada. No other competing interests were declared.
This article has been peer reviewed.
The authors have obtained patient consent.
Contributors: Adrian Budhram drafted the manuscript. Michael Silverman and Jorge G. Burneo edited the manuscript for intellectual content. All authors approved the final version to be published and agreed to act as guarantors of the work.
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

