


Patient 1
A 19-year-old Aboriginal woman presented with a three-week history of swollen neck glands, nausea, vomiting, chills and weight loss. On examination, she had bilateral, nontender, diffuse cervical lymphadenopathy. Oral examination revealed extensive dental caries and periodontal disease. The results of her laboratory workup are shown in Table 1. Both a lymph node aspiration and a bone marrow biopsy were nondiagnostic. The patient was scheduled for a surgical lymph node biopsy, but when she was seen one month later, the lymphadenopathy had resolved spontaneously. Based on these findings, she was diagnosed with Kikuchi–Fujimoto disease. At follow-up a year and a half later, she had no evidence of recurrence.
Results of laboratory investigations in two patients with Kikuchi–Fujimoto disease
Patient 2
Two years after the initial presentation of the first patient, her 19-year-old younger sister was assessed for a three-week history of swollen neck glands, night sweats, decreased appetite and weight loss. The sister also had severe dental and periodontal disease. A computed tomography (CT) scan of the neck showed severe bilateral lymphadenopathy, with the largest lymph node measuring 2.6 × 1.2 × 2.6 cm. Further CT scans showed moderate lymphadenopathy in the mediastinal, right hilar, axillary and pelvic regions. The results of a laboratory work-up are shown in Table 1. An excisional biopsy of a cervical lymph node and a bone marrow biopsy were done. By one month after presentation, the patient’s symptoms and signs had resolved, and she was discharged home. She was also diagnosed with Kikuchi–Fujimoto disease. At follow-up seven months later, she had a normal physical examination and her laboratory test results were normal.
Given that both siblings presented with an uncommon disorder at the same age, human leukocyte antigen (HLA) genotyping was done to examine whether shared immunogenetics may account for a predisposition to this disease. The results are shown in Table 1.
Pathology findings
In both sisters, the results of bone marrow examination were normocellular for all lineages and showed no evidence of myelodysplasia or features of a neoplastic infiltrate. Aspiration of a cervical lymph node in the first sister was nondiagnostic. In the second sister, an excisional biopsy showed geographic areas of necrosis containing apoptotic bodies and a striking degree of karyorrhexis with nuclear debris (Figure 1). Cells within the areas of necrosis were highly proliferative; more than 60% of the cells tested positive for the Ki-67 proliferation marker. Cellular material included a large number of histiocytes (CD68-positive and myeloperoxidase-positive), some with crescent-shaped nuclei, as well as large transformed lymphocytes (primarily CD8-positive T cells) (Figure 2). No neutrophils and only a few plasma cells were seen. Staining tests for organisms (periodic acid-Schiff reaction, Gömöri methenamine silver stain and Ziehl–Neelsen staining method) were negative. No morphologic evidence of malignancy was seen.
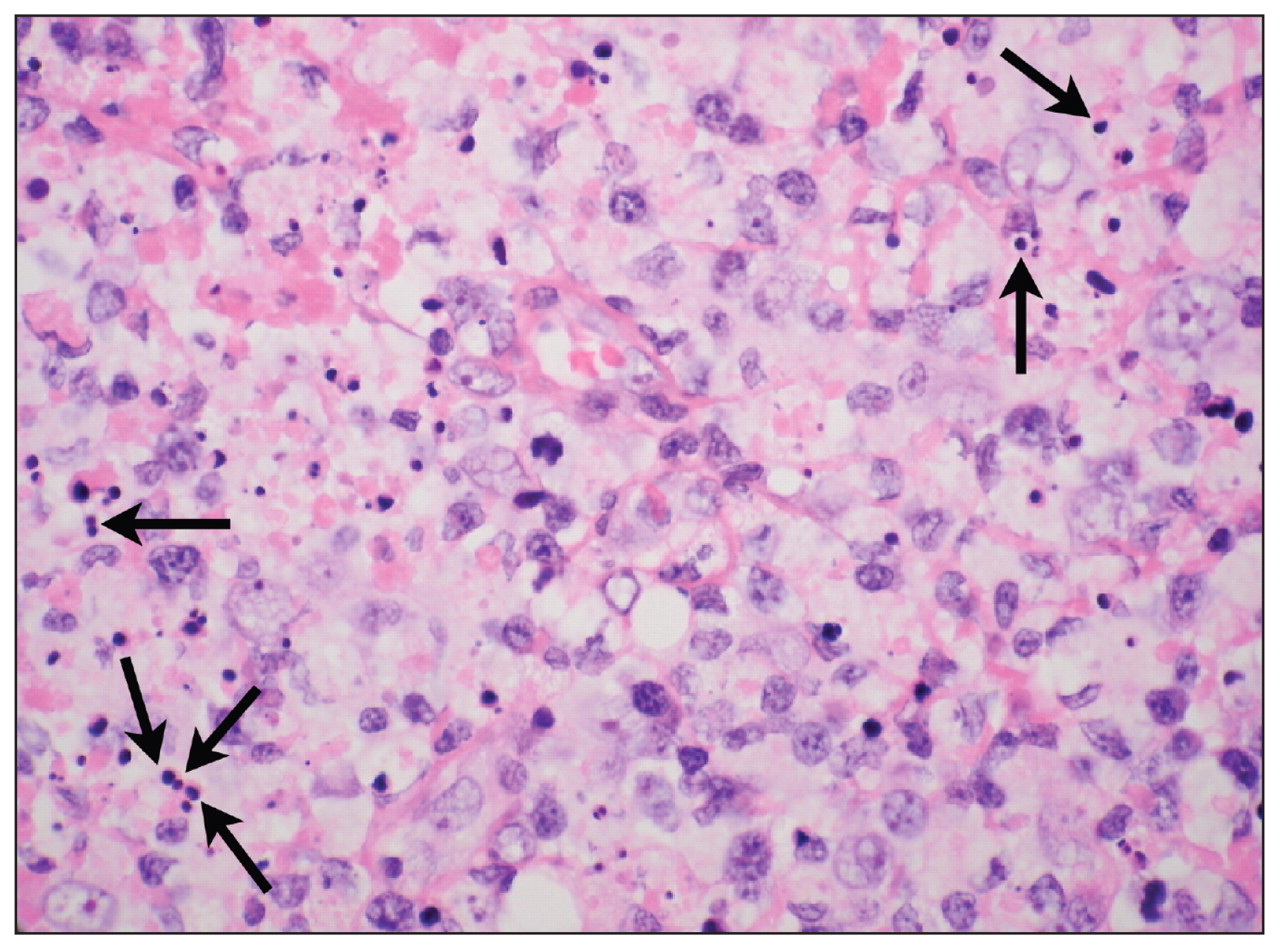
Microscopic view of lymph node biopsy specimen from Patient 2 (hematoxylin and eosin stain, magnification × 630), showing reactive histiocytes containing darkly stained, spherical karyorrhectic debris (arrows), a lack of neutrophils or malignant cells, and relatively few plasma cells.

Microscopic view of lymph node biopsy specimen from Patient 2 (CD68 stain, magnification × 200), emphasizing the numerous histiocytes or macrophages with brown staining.
Discussion
Kikuchi–Fujimoto disease is also known as Kikuchi disease or histiocytic necrotizing lymphadenitis. It was first described in Japan by Kikuchi and Fujimoto in 1972, and has now been reported in most regions of the world. Typical presentation includes fever, cervical lymphadenopathy and leukopenia, but may also include night sweats, nausea, vomiting and weight loss.1 Lymphadenopathy is most commonly cervical (79%), but can include any lymph node region and can be generalized (Box 1).1 The median age at presentation is 30 years. Most patients (70%) are young adults, but the disease can occur at almost any age.1,2 Reported case series support a female predilection with a male:female ratio ranging from 1:4 to 1:1.3.1,3 The disease is self-limiting, has no definitive treatment and spontaneously resolves within one to four months. The risk of recurrence is 3%–4% and can occur months to years after the initial diagnosis.4
Prevalence of the characteristics of Kikuchi–Fujimoto disease1
-
Lymphadenopathy (100%)
-
Leukopenia (43%)
-
Elevated erythrocyte sedimentation rate (40%)
-
Fever (35%)
-
Anemia (23%)
-
Rash (10%)
-
Weight loss (10%)
-
Fatigue (7%)
-
Joint pain (7%)
-
Night sweats, nausea, vomiting or diarrhea (< 7%)
The presentation of Kikuchi–Fujimoto disease closely mimics that of lymphoma and is frequently misdiagnosed as such. Patients often undergo extensive investigations for infectious, neoplastic and autoimmune conditions before a diagnosis of Kikuchi–Fujimoto disease is considered. The differential diagnosis for this disease is shown in Box 2.1,2 Diagnosis cannot be made using standard laboratory or diagnostic modalities; a lymph node biopsy is required. Histologic criteria for Kikuchi–Fujimoto disease are listed in Box 3.1,3,5
Differential diagnosis of cervical lymphadenopathy1,2
Infectious lymphadenitis
-
Herpes simplex virus
-
Epstein–Barr virus
-
Cytomegalovirus
-
Cat scratch disease (Bartonella henselae)
-
Group A streptococcus infection
-
Staphlyococcus aureus infection
-
Mycobacterium tuberculosis
-
Toxoplasmosis
-
Tularemia (Francisella tularenesis)
Neoplasia
-
Non-Hodgkin lymphoma
-
Hodgkin lymphoma
-
Metastatic carcinoma
Lymphadenitis of systemic lupus erythematosus
Kawasaki disease
Kikuchi–Fujimoto disease
Features of Kikuchi–Fujimoto disease on lymph node biopsy1,3,5
-
Erosion of lymphatic tissue
-
Circumscribed eosinophilic fibrotic necrotic foci with increased karyorrhexis
-
Histiocytes surrounding the necrotic foci
-
Nuclear debris
-
Increased lymphocytes (predominantly CD8-positive T cells)
-
Absence of granulocytes
-
Paucity of plasma cells
Kikuchi–Fujimoto disease has been found in association with various viruses, bacteria and parasites, but as of yet no clear causal relation has been shown.1,3,5 Kikuchi–Fujimoto disease shares many characteristics with systemic lupus erythematosus, including similar clinical, epidemiologic and histologic patterns. Patients who have had an episode of Kikuchi–Fujimoto disease may be at increased risk for systemic lupus erythematosus in the future, although the extent of this risk is unknown.6
Human leukocyte antigen genotyping
There are few reported instances of familial Kikuchi–Fujimoto disease or familial associations in the published literature.4,7–9 Human leukocyte antigen (HLA) genotyping for familial Kikuchi–Fujimoto disease has been reported in only one paper.4 The sisters described in that case share some HLA genotypes as the sisters described here; both of our patients tested positive for HLA-Cw7 and the older sister (Patient 1) also tested positive for HLA-B35. Neither of these genotypes is known to be associated with an increased risk of nonfamilial Kikuchi–Fujimoto disease.7
Shared environmental exposure may also explain the coincident diagnosis of Kikuchi–Fujimoto disease in these sisters. Both siblings were from a remote Northern Ontario reserve and were Aboriginal. Given that they both had very poor dentition with associated periodontitis, perioral microorganisms may have triggered the development of this disease. However, no association between oral infection and development of Kikuchi–Fujimoto disease has been reported, so this hypothesis remains to be investigated.
Conclusion
Although uncommon, Kikuchi–Fujimoto disease should be considered in the differential diagnosis of cervical lymphadenopathy. In particular, it can be mistaken for malignant lymphoma. The contribution of genetic or environmental factors to the development of Kikuchi–Fujimoto disease remains unknown. Although the disease is self-limiting, long-term follow-up is important to monitor for recurrence of the disease or development of autoimmune disorders, such as systemic lupus erythematosis.
-
Kikuchi–Fujimoto disease is an uncommon and sometimes familial disorder that should be considered in the differential diagnosis of cervical lymphadenopathy.
-
Typical presentation includes fever, leukopenia and cervical lymphadenopathy.
-
Although Kikuchi–Fujimoto disease is self-limiting and no definitive treatment exists, lymph node biopsy is required to rule out malignancy.
-
Patients should be followed closely because of increased risk for recurrence and for systemic lupus erythematosus.
Key points
Footnotes
-
This article has been peer reviewed.
-
Competing interests: None declared.
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

