


Neuroendocrine tumours (NETs) are a unique group of malignant growths, best known for their ability to secrete bioactive peptides, which may cause symptoms such as flushing and diarrhea. Although NETs traditionally have been considered uncommon, a recent population-based study reported that the incidence of NETs in Canada had risen markedly from 2.48 to 5.86 per 100 000 per year between 1994 and 2009.1 For reference, the incidence of NETs is now similar to that of cervical cancer according to the most recent Canadian Cancer Society statistics.2
Because NETs are perceived to be uncommon and may be nonspecific in their presentation, delays in diagnosis are frequent. A recent international survey of 1928 patients with NETs reported a mean delay of 52 months between symptom onset and diagnosis; patients see an average of six different health care providers before receiving the correct diagnosis.3 Unfortunately, when patients are ultimately diagnosed with a NET, many will have metastases; population-based studies have reported that 21% of patients are found to have metastases at the time of diagnosis, 1,4 whereas proportions as high as 56%–69% have been reported in retrospective chart reviews.5,6
This review summarizes the classification, presentation, diagnostic workup and treatment of NETs with the aim of helping generalists to facilitate timely diagnosis and referral. Diagnostic recommendations made in this review are based on consensus expert opinion, whereas recommendations for systemic treatment are largely based on phase three randomized controlled trials (RCTs), which compare new treatments with standard treatment. All recommendations are consistent with current Canadian national guidelines.7,8 Box 1 summarizes the evidence supporting this review.
What are neuroendocrine tumours?
Neuroendocrine tumours are malignant growths that arise from neuroendocrine cells. They most commonly occur in the gastrointestinal tract (48%), lung (25%) and pancreas (9%), but may also develop in many other organs, including the breast, prostate, thymus and skin.1 Neuroendocrine cells have the capability to produce hormones, such as serotonin,9 which can result in symptoms such as flushing and diarrhea, as well as other proteins (e.g., chromogranin A), which serve as biomarkers for NETs.10–12 Neuroendocrine tissues also tend to express somatostatin receptors on their cell surfaces. Therefore, somatostatin analogues can be useful for diagnostic imaging and treatment.13,14
How are neuroendocrine tumours classified?
There are three clinicopathologic features that drive the biological behaviour of NETs: grade, differentiation and stage.
Grade
Histologic grade reflects the biological aggressiveness of the neoplasm. There are two main features of grade: the Ki67 index, which measures the percentage of cancer cells that stain positive for Ki67, a marker of cell proliferation (Figure 1) and the mitotic rate, which documents the number of mitoses per 10 high-power microscopic fields.15–17 The Canadian National Expert Group on NETs recently endorsed the grading system from the World Health Organization (Table 1).7,8,18,19 The importance of accurate grade cannot be overemphasized as it is the key determinant of prognosis.4,7,8 Based on data from the Surveillance, Epidemiology and End Results (SEER) Program (United States National Cancer Institute) database, the median survival for patients with a grade 1, 2 or 3 tumour is 124 months, 64 months and 10 months, respectively.4
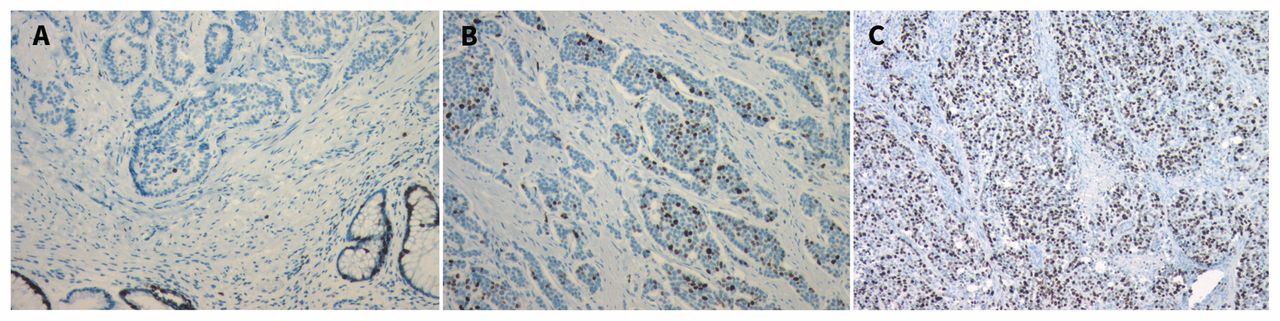
Pathology specimens of tissue from a neuroendicrine tumour with staining for Ki67 (a nuclear protein involved with cell proliferation). MIB-1 is an immunoglobulin G (IgG) antibody directed against Ki67 that can be used to identify the percentage of cells undergoing active cell proliferation. (A) Ki67 index < 3%. (B) Ki67 index 3%–20%. (C) Ki67 index > 20%. Images provided by Dr. Corwyn Rowsell (St. Michael’s Hospital, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ont.).
Differentiation
Differentiation refers to how closely the neoplastic cells resemble their nonneoplastic counterparts in the tissue from which they arose. Well-differentiated cancer cells closely resemble nonneoplastic cells, whereas poorly differentiated cancer cells do not. In general, low-grade tumours (grades 1 and 2) are well-differentiated and high-grade tumours (grade 3) are poorly differentiated.15
Stage
Stage refers to extent of tumour spread throughout the body and, as for other cancers, the Tumor Node Metastasis (TNM) system is used. The Canadian National Expert Group on NETs recently endorsed the staging groupings described in the 7th edition of the American Joint Committee on Cancer Staging Manual. 7,8,21 However, for practical purposes, NETs can be considered as either early stage (completely resectable) or advanced stage (either locally advanced and unresectable or metastatic).
Nomenclature
The German pathologist Sigfried Oberndorfer coined the term “Karzinoide” in 1907, meaning carcinoma-like, to describe small benign-appearing tumours of the small intestine.22
The term “carcinoid” is no longer recommended, because it fails to convey the malignant potential that most NETs harbour. The term is also confusing, because it promotes the misconception that all NETs produce the carcinoid syndrome, when most do not.5,6,23–26 Currently, the term “neuroendocrine tumour” is preferred to describe grade 1 and 2 tumours, whereas the term “neuroendocrine carcinoma” is used to describe grade 3 tumours.
How do neuroendocrine tumours present?
Neuroendocrine tumours may be found as an incidental finding or may be suspected from clinical symptoms. When NETs cause clinical symptoms from secreted hormones, they are termed “functioning” (Table 2). Most NETs do not produce a biologically active hormone and are termed “nonfunctioning.”5,6,23–26 Both functioning and nonfunctioning NETs tend to present late with nonspecific symptoms that are often attributed to an alternative diagnosis (Table 3). In the aforementioned international survey of patients with confirmed NETs, the most common initial diagnoses provided to patients included gastritis, irritable bowel syndrome, anxiety, inflammatory bowel disease, asthma and menopause.3
Some symptoms may suggest the diagnosis and location of a NET. Small intestinal NETs may cause extensive fibrosis, which result in recurrent abdominal pain secondary to small bowel obstruction or mesenteric ischemia. In a retrospective study of 121 consecutive patients with midgut NETs who underwent surgical resection, marked mesenteric fibrosis and intestinal ischemia were found in 65% and 38% of patients, respectively.28 Bronchopulmonary NETs tend to present with centrally located lesions that may result in bronchial obstruction, recurrent obstructive pneumonitis, cough and hemoptysis.24–26 Furthermore, bronchopulmonary NETs may be a source of ectopic adrenocorticotrophic hormone (ACTH) production; clinicians should consider this diagnosis in otherwise unexplained Cushing syndrome.25,29–31
Carcinoid syndrome
Carcinoid syndrome, characterized by flushing, diarrhea and valvular heart disease, occurs when hormones produced by NETs reach systemic circulation. This happens most commonly after liver metastases develop, allowing for bypass of hepatic metabolism that may inactivate the hormones. Retrospective cohort studies report that carcinoid syndrome occurs in 6%–13% of patients with pathologically confirmed gastrointestinal NETs and in less than 1% of patients with bronchopulmonary NETs.24,25,32–34 Hindgut tumours (distal colon and rectum) are typically hormonally silent and do not cause carcinoid syndrome.34
Evidence used in this review
We searched MEDLINE, Embase, Google Scholar and the Cochrane Database of Systemic Reviews for articles relating to the diagnosis, classification and management of neuroendocrine tumours. Search terms included “neuroendocrine tumors,” “neuroendocrine neoplasms,” “carcinoid tumors,” “islet cell tumors” and “bronchopulmonary neuroendocrine tumors.” We then searched for abstracts from major international oncology conferences to identify any recently completed studies. The reference lists of all relevant articles were reviewed for studies not captured in the original search.
Valvular heart disease secondary to fibrotic plaques has been reported in 8%–56% of patients with carcinoid syndrome in retrospective cohort studies.23,35–37 Because 43% of patients in these studies were asymptomatic, screening echocardiography in this group is important to allow timely intervention of valvular disease.36
Carcinoid crisis is an acute life-threatening presentation of carcinoid syndrome characterized by profound flushing, bronchospasm and rapidly fluctuating blood pressure. It may be precipitated by induction of anesthesia or palpation, ablation or embolization of a NET.38 Therefore, patients should be given a somatostatin analogue before any anesthetic or tumour manipulation.7,8
How are neuroendocrine tumours diagnosed?
The diagnosis of a NET requires a coordinated multidisciplinary effort involving medical oncologists, surgeons, interventional radiologists and pathologists. Results from pathology testing, hormonal testing, and diagnostic and functional imaging are integrated to form a comprehensive diagnostic picture.
Pathology
Obtaining tissue for pathology testing is mandatory for the diagnosis of NETs. When surgical resection is not an option, core needle biopsy is preferred over fine needle aspiration to allow full assessment of the tumour architecture.7,8
Syndrome-specific biochemical testing
For patients presenting with symptoms of a functioning NET, biochemical testing should be targeted to the specific syndrome (Table 2). For patients presenting with a small intestinal mass or with features of carcinoid syndrome, a 24-hour urine 5-hydroxyindole acetic acid (5-HIAA) test should be ordered. This test has a reported sensitivity of 35%–73%.12,39,40 Patients must refrain from eating serotonin-rich foods (e.g., bananas, pineapples, avocados, kiwi fruits or nuts) for at least three days before testing to prevent false-positive results.
Nonsyndrome-specific biochemical testing
Chromogranin A is the diagnostic biomarker of choice for NETs.10,41 It has a high sensitivity (53%–91%) but low specificity (< 50%).10,42,43 The most common reasons for false elevations include proton-pump inhibitors, renal insufficiency, adenocarcinomas and severe arterial hypertension.10,44
Diagnostic imaging
There are two general categories of diagnostic imaging modalities that are used in combination for the diagnosis of NETs.
The first modality is standard cross-sectional imaging with computed tomography (CT) or magnetic resonance imaging (MRI). Canadian consensus guidelines recommend that all patients with a suspected NET should receive CTs of the chest, abdomen and pelvis for staging, and consideration of an MRI of the liver or pancreas in cases where further definition is required.7,8
The second modality is functional imaging, which takes advantage of the overexpression of somatostatin receptors commonly seen in NETs. Radiolabelled somatostatin analogues are administered intravenously, concentrate in NETs, and the emitted radiation is detected to localize tumours. 111Indium (In)-labelled pentetreotide is the most commonly used radiotracer in Canada. A recent meta-analysis of molecular imaging methods for NETs reported that 111In-labelled pentetreotide somatostatin receptor scintigraphic imaging had a sensitivity of 46%–100% for abdominal NETs, 46%–83% for pancreatic NETs and 71% for bronchial NETs.45
A newer radioisotope, 68Ga (gallium), is a positron emitter that also can be linked to somatostatin analogues and localized with positron emission tomography (PET)/CT imaging.46 In a recent prospective single-centre study in the US, 78 patients with NETs underwent imaging with both 111In-labelled pentetreotide scans and 68Ga-DOTATATE PET/CT.47 The sensitivity of 68Ga-DOTATATE PET/CT compared with imaging using 111In-labelled pentetreotide was 96% (95% confidence interval [CI], 86%–100%) versus 72% (95% CI, 58%–84%), respectively. Although access to 68Ga-based imaging is currently limited to subspecialty centres in Canada, it is expected to become widely available soon.
It is important to note that somatostatin analogue–based imaging modalities have poor sensitivity in poorly differentiated cancer, because the cells often lack somatostatin receptors. Therefore, poorly differentiated tumours are better imaged with a fluorodeoxyglucose (18FDG) PET/CT scan, which localizes tumours by uptake of radiolabelled glucose molecules (Figure 2).48
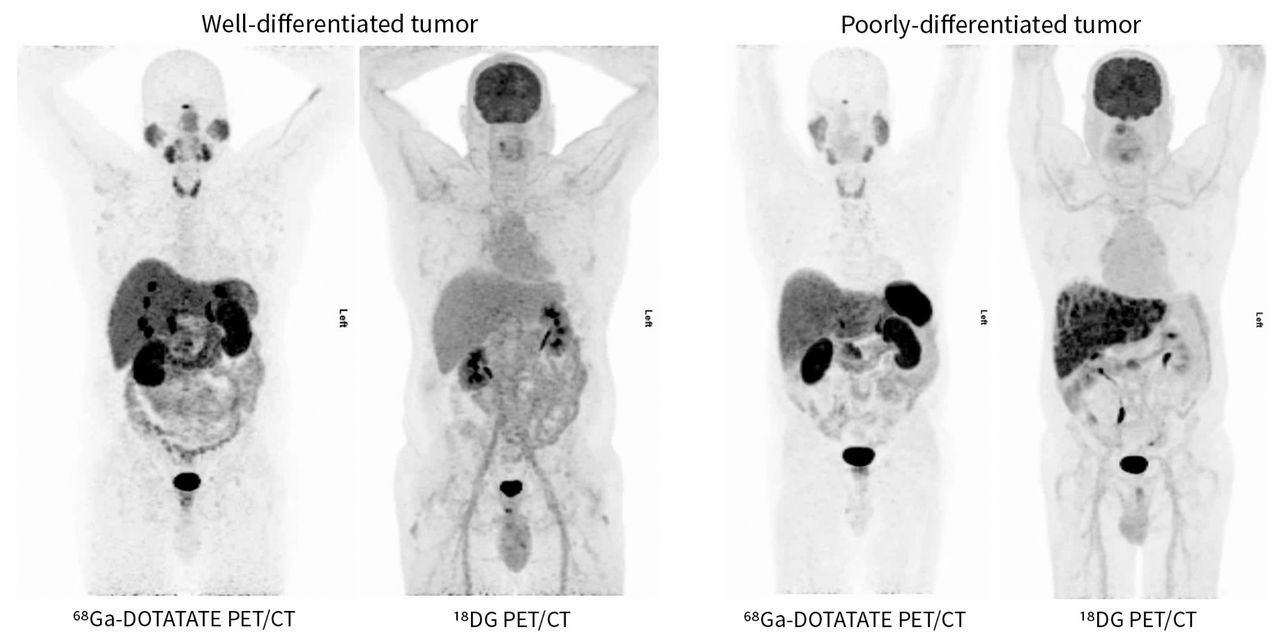
Comparison of 68Ga-DOTATATE PET/CT and 18FDG PET/CT for detection of poorly and well-differentiated neuroendocrine tumours. 68Gallium radiolabelled somatostatin analogue PET/CT is more sensitive for identifying well-differentiated tumours in which the somatostatin receptor expression is preserved on neuroendocrine cells. 18FDG PET/CT is more sensitive for identifying poorly differentiated tumours in which the somatostatin receptor expression has been lost. Please note that the comparison images are from from different patients to illustrate differences in detail by modality based on the differentiation of the tumour. Images provided by Dr. David Chan (Department of Medicine, Sunnybrook Health Sciences Centre, University of Toronto, Toronto, Ont.). FDG = fluorodeoxyglucose, PET/CT = positron emission tomography–computed tomography.
Endoscopic imaging
Endoscopic ultrasonography is the most sensitive test for the diagnosis of pancreatic NETs (sensitivity 82%–93%).49,50 Endoscopic ultrasonography is particularly useful for identifying tumours less than 2 cm in size and for the localization of insulinoma, 51 and often is employed intraoperatively for this purpose.
How are neuroendocrine tumours treated?
Individualized treatment plans are formulated based on tumour factors such as site, stage, grade, differentiation and symptoms, and patient factors such as age and comorbidities.
Localized disease
Surgery
An appropriate oncologic operation focused on margin-negative resection and adequate lymphadenectomy is the only curative treatment modality to date. At the time of resection, it is crucial that the surgeon carefully inspect for synchronous lesions. A retrospective single-centre study involving 691 patients with midgut NETs identified multiple synchronous primary tumours in 22% of patients, with a range of 2 to 26 tumours.23
Adjuvant therapy (chemotherapy after surgery)
No adjuvant therapy has been proven to improve cure following surgery for NETs. However, based on extrapolation of small cell lung cancer data, consensus guidelines recommend that adjuvant platinum-based chemotherapy be considered for patients with fully resected, poorly differentiated cancers.7,8
Metastatic disease
Observation
In select patients with low-volume, asymptomatic, nonfunctional metastatic disease, observation with expectant management and serial diagnostic imaging is appropriate; historically, many patients remain well without disease progression for years.7,8
Somatostatin analogues
Somatostatin analogues are a cornerstone of the treatment of NETs. Two long-acting somatostatin analogue preparations are available: octreotide long-acting repeatable and lanreotide. Somatostatin analogues were used initially in patients with secretory symptoms only, but two phase 3 RCTs published in 2009 and 2014 showed their anti-proliferative effect.52,53 In the PROMID trial, 90 patients with well-differentiated metastatic NETs were randomly assigned to either placebo or octreotide. Octreotide long-acting repeatable was associated with improved time to progression (hazard ratio [HR] 0.34, 95% CI 0.20–0.59).52 In recently updated survival analyses, no effect was seen on median overall survival (84.7 v. 83.7 mo), but this was felt to be due to the crossover of most patients from placebo to treatment.54 Similarly, in the CLARINET trial, 204 patients with moderate to well-differentiated nonfunctioning NETs were randomly assigned to lanreotide or placebo. Lanreotide was also associated with improved progression-free survival (HR 0.47, 95% CI 0.30–0.73).54
Adverse effects of using somatostatin analogues include diarrhea, abdominal pain, nausea, vomiting and hyperglycemia.54 Additionally, patients treated with somatostatin analogues have an increased risk of cholelithiasis and biliary sludge development; therefore, prophylactic cholecystectomy should be considered for patients starting long-term treatment with somatostatin analogues.7,8,55,56 However, this recommendation has never been evaluated in a prospective study and is based on retrospective studies that showed high rates of cholelithiasis (52%–63%) and modest rates of symptomatic gallbladder disease (7%–15%).55,56
Surgery
Surgery plays an important role even in the setting of metastatic disease. Resection of the primary, if located in the small bowel, is usually undertaken to prevent obstruction later, particularly for low-grade tumours with good prognosis. The findings of retrospective population-based studies suggest that this may also improve survival.57,58 In the presence of large-volume metastatic disease, surgical cytoreduction can be undertaken to improve control of secretory symptoms that may be hard to control with somatostatin analogue alone; there is also evidence from retrospective studies that this may improve survival.59–61
Molecularly targeted biologic therapy
Everolimus is an oral mammalian target of rapamycin (mTOR) inhibitor that is thought to act by inhibiting cell proliferation, angiogenesis and cell metabolism. Everolimus has proven progression-free survival benefit in well-conducted phase 3 RCTs in gastrointestinal and lung NETs,62–64 and pancreatic NETs.65,66 In the RADIANT-4 phase III RCT involving 302 patients with advanced progressive nonfunctional lung or gastrointestinal NETs, everolimus was associated with a 52% reduction in the risk of progression or death (HR 0.48, 95% CI 0.35–0.67) compared with placebo. Similarly, in the RADIANT-3 phase 3 RCT involving 410 patients with advanced low- or intermediate-grade pancreatic NETs, everolimus was associated with a 65% reduction in the risk of progression or death (HR 0.35, 95% CI 0.27–0.45) compared with placebo.
Important adverse effects of everolimus include stomatitis, diarrhea, fatigue, pneumonitis, hypophosphatemia, hyperlipidemia and hyperglycemia.
Sunitinib is a multitargeted tyrosine kinase inhibitor that is thought to act by inhibiting angiogenesis. Treatment efficacy for pancreatic NETs was shown in a well-conducted phase 3 RCT involving 171 patients with well-differentiated pancreatic NETs. Compared with placebo, sunitinib was associated with improved progression-free survival (HR 0.42, 95% CI 0.26–0.66) and overall survival (HR 0.41, 95% CI 0.19–0.89).67
Important adverse effects of sunitinib include diarrhea, nausea, vomiting, fatigue, changes to hair colour, hypertension, palmar–plantar erythrodysethesia (hand–foot syndrome) and hypothyroidism.
Peptide receptor radionuclide therapy
Peptide receptor radionuclide therapy involves attaching a therapeutic radiolabel to a somatostatin analogue that then concentrates in neuroendocrine tissue. The radiolabel, most commonly177Lutetium (177Lu), can then deliver local cytotoxic radiation to the NET. It is administered intravenously every eight weeks for four doses. In the recently reported NETTER-1 phase 3 RCT, compared with placebo, patients who received peptide receptor radionuclide therapy after progression on somatostatin had a significantly increased progression-free survival (median overall survival not yet reached v. 8.4 mo, p < 0.001).68
Important adverse effects of peptide receptor radionuclide therapy include renal dysfunction, nausea, vomiting and suppression of bone marrow.
Chemotherapy
There are no high-quality RCTs guiding chemotherapy treatment for metastatic poorly differentiated cancers. Most evidence is extrapolated from the literature on small cell lung cancer. Similar to small cell lung cancer, there is usually a dramatic and rapid response to chemotherapy, but subsequent relapse is common, with median overall survival of less than 1 year.69,70
Unanswered questions
Several unanswered questions remain. What are the best predictive and prognostic markers for patients with neuroendocrine tumours? What is the optimal follow-up strategy for patients with neuroendocrine tumours? How should the multiple new therapeutic options best be sequenced and/or combined? How do the new therapeutic options affect patient quality of life and survivorship?
Conclusion
Incidence and prevalence of neuroendocrine tumours are increasing. Awareness of this heterogeneous entity may reduce delay in diagnosis and facilitate expert multidisciplinary care. Treatment options are rapidly expanding; therefore, many patients could have long periods of well-controlled symptoms and a better quality of life.
KEY POINTSNeuroendocrine tumours are increasing in both incidence and prevalence in Canada.
Most patients present with non-specific symptoms such as cough, abdominal pain, bloating and weight loss; few patients present with all features of the classical carcinoid syndrome characterized by flushing, diarrhea and valvular heart disease.
Clinicians should have a high index of suspicion to ensure timely diagnosis and referral to multidisciplinary subspecialty centres.
Treatment options are rapidly expanding and many patients with neuroendocrine tumours may have extended survival periods with well-controlled symptoms and a better quality of life.
Footnotes
Competing interests: Calvin Law and Simron Singh received honoraria from Ipsen Canada as members of their advisory boards. David Chan received speaker fees from Ipsen Australia. David Chan and Simron Singh received nonfinancial support from Novartis. Calvin Law received speaker fees and travel support from Amgen Canada and Novartis Oncology, and honoraria from Baxalta, Ipsen Canada and Novartis Oncology as a member of their advisory boards. He also received nonfinancial support from Amgen Canada. Simron Singh received travel support, speaker fees and nonfinancial support from Novartis Oncology, and nonfinancial support from Pfizer. He also received a grant from Novartis and nonfinancial support from Ipsen Canada. No other competing interests were declared.
Contributors: All authors contributed substantially to the conception and design of the review. Michael Raphael performed the original literature search and drafted the first version of the manuscript. David Chan, Calvin Law and Simron Singh revised the manuscript critically for important intellectual content. All of the authors approved the final version to be published and agreed to be accountable for all aspects of the work.
This article was solicited and has been peer reviewed.
References
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

