


Prion diseases are rare, lethal, untreatable degenerative brain disorders of humans and animals. They are linked to conversion of a specific brain protein from its normal form (PrPC) into a misfolded, pathologic form (PrPSc) and are transmissible between individuals by a novel protein-based mechanism.1 Because there is no convincing evidence for causation by a conventional microbial pathogen, and because even a minute quantity of exogenous material containing PrPSc can initiate a cascade of PrPC conversion in a new host in a manner reminiscent of infection, the term “prion” was coined to denote the implicated proteinaceous infectious particle.1
Population-based estimates of mortality related to human prion disease fall in the range of one to two per million population annually.2 Sporadic Creutzfeldt–Jakob disease (CJD), which occurs without an identifiable genetic or infectious cause, accounts for 85%–95% of all cases.2 Mutations in the chromosomal gene, PRNP, that encodes PrPC cause most of the remaining 5%–15% of deaths,3 with less than 1% attributable to infectious transmission through medical procedures or the zoonotic route.4,5 Regardless of etiology, all prion diseases are considered potentially transmissible, which is why these diseases have sustained public health concern for the past several decades, despite their rarity.6 The Public Health Agency of Canada conducts national surveillance of human prion diseases, with the support of formal systems for provincial and territorial reporting and national notification.7
For the clinician managing a case of suspected prion disease, timely, accurate diagnosis is particularly important, and there is often an urgency to identify potential risks of infectious transmission in the health care setting. With this in mind, and given that sporadic CJD accounts for most of the diagnostic effort devoted to human prion diseases, in this article we review the diagnostic usefulness of immunoassays of proteins in cerebrospinal fluid, focusing on 13 published studies of the proteins tau, 14-3-3 and S100B in cerebrospinal fluid (Box 1).
Evidence used in this review
We assessed published evidence on the diagnostic accuracies of the three cerebrospinal fluid protein markers most widely used in diagnosis of sporadic Creutzfeldt–Jakob disease (14-3-3, tau and S100B proteins). We searched PubMed using the following search profile: (“14-3-3”[All fields] OR “tau”[All fields] OR “S100B”[All fields]) AND (“CJD”[All fields] OR “Creutzfeldt-Jakob”[All Fields]) AND (“CSF”[All fields] OR “cerebrospinal”[All Fields]). We selected appropriate prospective diagnostic studies and reviewed 13 conducted by centres in 15 different countries. See details of reviewed studies in Appendix 1, available at www.cmaj.ca/lookup/suppl/doi:10.1503/cmaj.130720/-/DC1.
When may sporadic CJD be suspected?
The process of differential diagnosis8 can be difficult to apply to any rare disease. Unfamiliarity may impede the generation of diagnostic hypotheses, objective diagnostic opinion may be elusive, and criteria for whether and when to order supporting investigations may be unclear. In the case of sporadic CJD, heterogeneity and a broad differential add to the diagnostic challenge. Classic sporadic CJD is characterized by the following clinical pattern: sudden onset of inexorable, rapidly progressive dementia (average duration about 4 mo) in a patient over 50 years of age, with or without other prominent features such as myoclonus, ataxia, visual disturbances, psychiatric changes, aphasia, other motor symptoms and akinetic mutism in later stages.9 However, only a subset of presentations faithfully display these classic features, and the clinician must also be prepared to consider the diagnosis among patients with varying presentation, evolution or age of onset,10–12 and as part of a long list of non-CJD conditions that can also present as subacute encephalopathy.13 These differential diagnoses, of which many are far more common than sporadic CJD, span much of the spectrum of human disease, with vascular, infectious, inflammatory, autoimmune, neoplastic, degenerative, toxicologic, psychiatric, metabolic or other causes. Furthermore, some are treatable and must not be missed.14 The non-CJD conditions encountered during national CJD surveillance in Canada range across this broad spectrum.15 Murray offers an excellent overview of these “CJD mimics,” including recommendations on how to organize the process of differential diagnosis.13
Which investigations most usefully support the diagnostic process?
Diagnostic criteria for sporadic CJD stipulate detection of PrPSc as the gold standard.16 However, even with promising recent advances,17 current technology cannot routinely detect the trace levels of PrPSc in standard clinical specimens such as blood, urine or cerebrospinal fluid. Thus, diagnostic verification requires neuropathologic examination to detect PrPSc in brain tissue, which, despite occasional indications for brain biopsy,18 almost always takes place postmortem.16 In the living patient, diverse information from electroencephalography (EEG), brain imaging (e.g., magnetic resonance imaging [MRI]), biochemistry and genetics must be considered in addition to clinical presentation and history. This approach has been formalized in the World Health Organization’s case definitions for sporadic CJD, on which national surveillance of CJD in Canada is directly based (Appendix 1, available at www.cmaj.ca/lookup/suppl/doi:10.1503/cmaj.130720/-/DC1).7,19,20
Characteristic EEG findings (i.e., periodic triphasic sharp-wave complexes at about 1 Hz) are a hallmark of sporadic CJD but lack diagnostic sensitivity, with reported sensitivities of about 60% and even 30%.10,12 Findings on MRI (i.e., high signal in caudate nucleus or putamen with diffusion-weighted imaging or fluid-attenuated inversion recovery sequences) have proven useful in expert centres, especially in combination with EEG or analysis of cerebrospinal fluid markers.20–22 However, observer dependence and variability among disease subtypes can complicate the application of MRI.21,23 The most widely used supporting investigations are immunoassays for certain protein markers that are markedly elevated in lumbar cerebrospinal fluid in most affected patients (Box 2).24,25
How to obtain services for cerebrospinal fluid testing in Canada
A panel for cerebrospinal fluid markers for sporadic Creutzfeldt–Jakob disease, including 14-3-3, tau and S100B proteins, is available to Canadian clinicians at no cost through the Public Health Agency of Canada’s Prion Laboratory Section, National Microbiology Laboratory, 1015 Arlington St., Winnipeg MB R3E 3R2. Samples must be taken ante mortem, should be frozen as soon as possible after collection, and must be optically clear and nonxanthochromic. The sample should be shipped on dry ice as per Transportation of Dangerous Goods Regulations for UN 3373, Biological substance, Category B. For additional information, see the Guide to Services of the National Microbiology Laboratory (www.nml-lnm.gc.ca/guide2/index-eng.htm) or contact the laboratory at 204 789-6078 or nml.cjd{at}phac-aspc.gc.ca. Information about human prion diseases and diagnostic testing may also be obtained by contacting the Canadian Creutzfeldt–Jakob Disease Surveillance System at 888 489-2999 or cjdss{at}phac-aspc.gc.ca.
The most commonly used cerebrospinal fluid markers are 14-3-3 proteins, the microtubule-associated protein tau and the S100B protein, for which many studies have reported estimates of diagnostic sensitivity and specificity in the range 0.80–0.90 (Appendix 1).26,27 However, risks associated with false diagnoses have prompted cautions that these tests should be ordered only in an appropriate clinical context, that is, after the broad initial differential has been substantially narrowed.26,28 Although such caution is reasonable, application can be problematic because, in practice, the number of competing diagnoses at the time tests are ordered can vary widely from case to case. It may also be important (e.g., for reasons of hospital safety) to expedite testing before results from other investigations are available or before the patient’s disease has fully evolved.
Fortunately, such circumstances are well-suited to a structured, evidence-based decision-making approach, designed to systematically reduce diagnostic uncertainty while mitigating common cognitive biases.8,29 A brief review of this approach is presented in Appendix 1. In this review, we summarize the data of 13 published studies within such a framework.
How might an evidence-based interpretive framework be constructed?
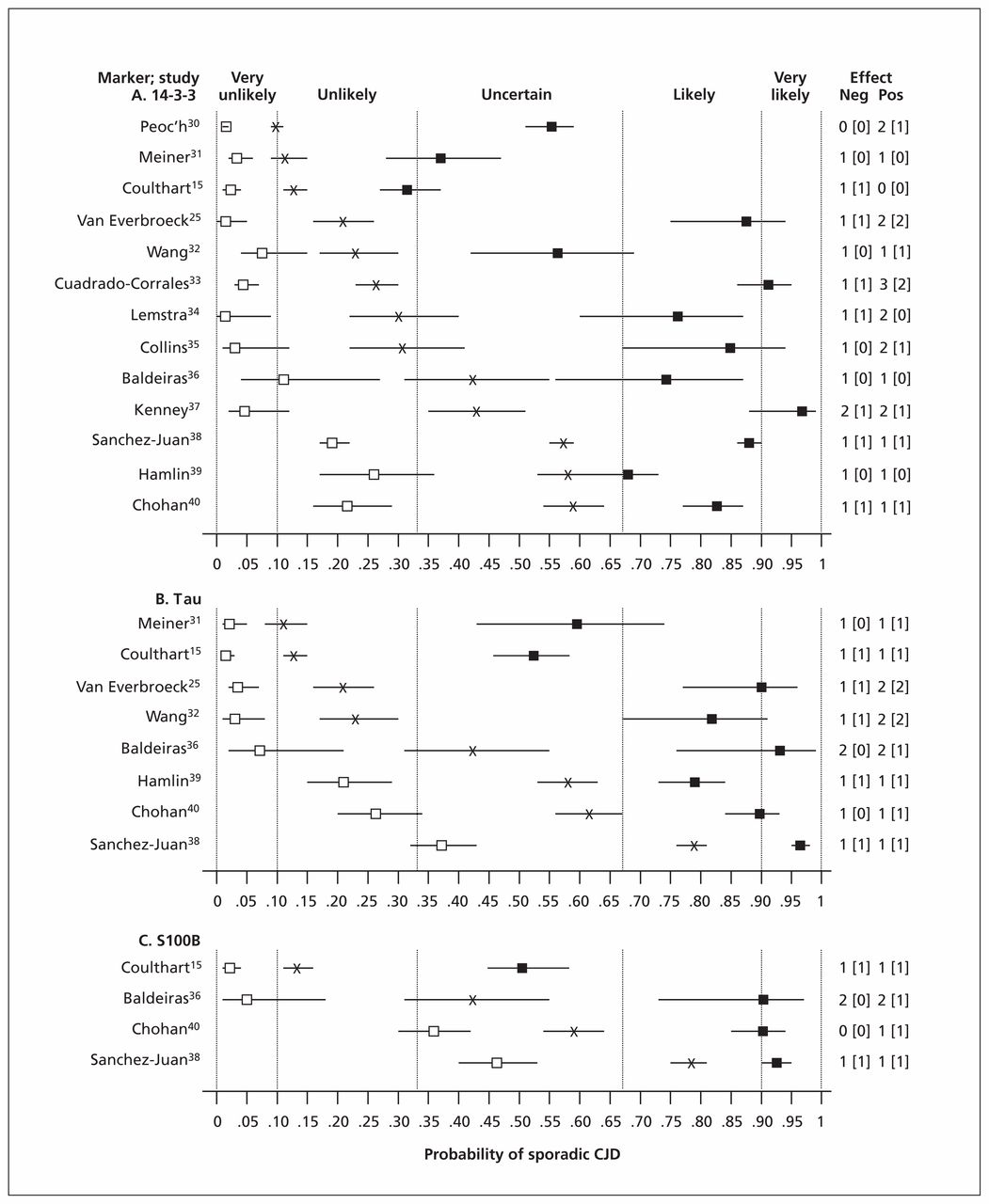
To estimate the effects of the results of cerebrospinal fluid testing on diagnostic uncertainty, we defined pretest probabilities as prevalences of sporadic CJD observed in the reviewed studies. Using Bayes theorem and study-specific estimates of test accuracy, we then calculated post-test diagnostic probabilities for the presence of sporadic CJD with both positive and negative test results (Figure 1). There was wide variation in prevalence, from 0.10 (95% confidence interval [CI] 0.09–0.11) to 0.79 (95% CI 0.76–0.81). As expected, posttest probabilities also varied substantially. Given the close similarity of laboratory methodology among studies, the variations in both prevalence and posttest probability are likely attributable to differences in composition of patient populations.
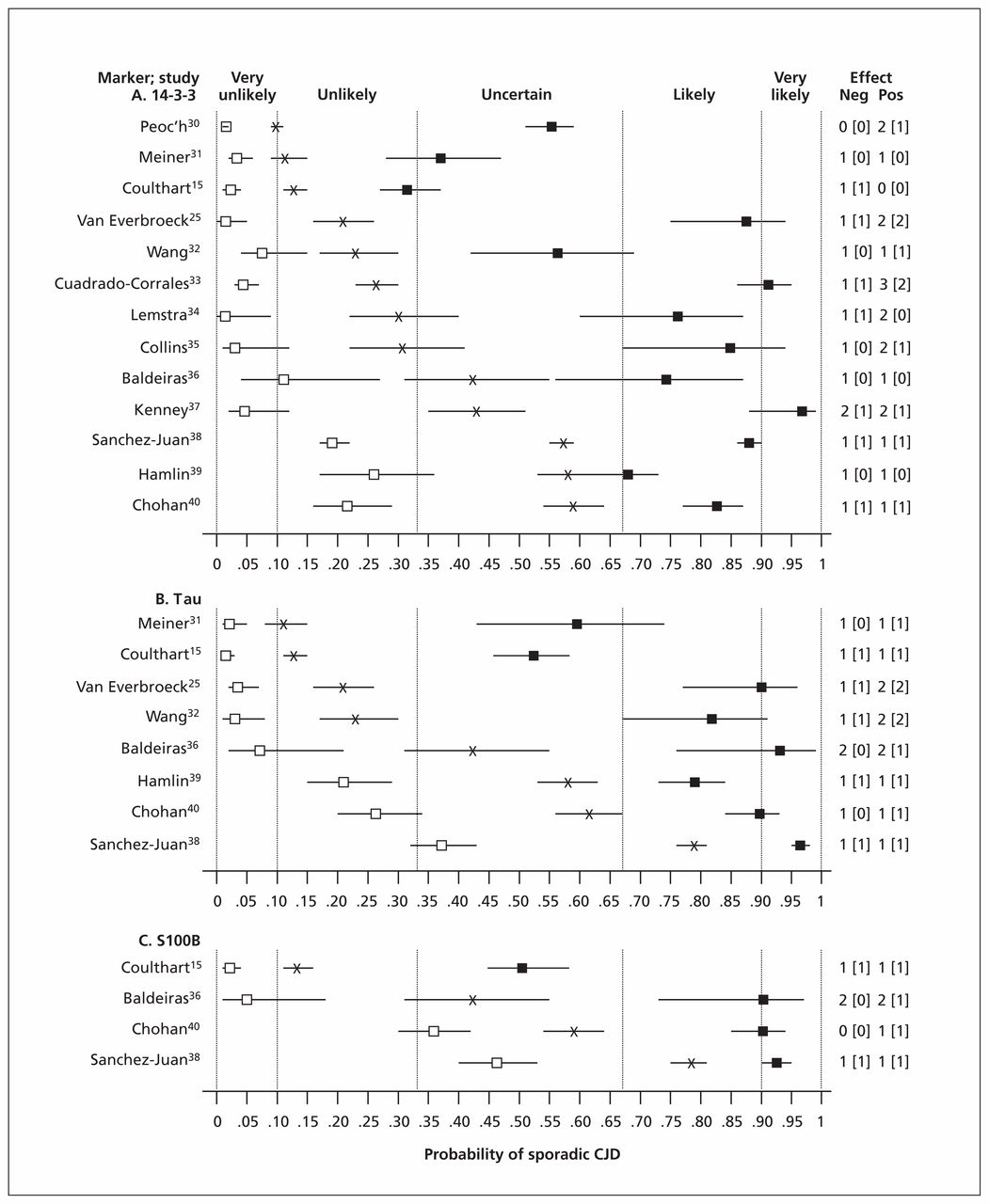
Effects on diagnostic probabilities of results of cerebrospinal fluid protein tests for 14-3-3 (A), tau (B) and S100B (C) in sporadic Creutzfeldt–Jakob disease (CJD). X: pretest probability (sporadic CJD prevalence); closed and open squares: posttest probability for positive or negative test results, respectively; horizontal bars: 95% confidence interval (CI); vertical dashed lines: diagnostic opinion category (Box 2); numbers at right: size of effect on diagnostic probability in terms of category differences, for negative (Neg) and positive (Pos) test results; numbers in square brackets: effect size with strict application of 95% CI.
Clinical interpretation of these findings is facilitated by the insights of Medow and Lucey41 (Box 3), who observed on theoretical grounds that when test sensitivity and specificity are in the range 0.80–0.90 (as with many good tests), test results tend to modify diagnostic probabilities by one step between five categories defined intuitively as very unlikely (< 0.10), unlikely (0.10–0.33), uncertain (0.33–0.67), likely (0.67–0.90) and very likely (> 0.90). Thus, as shown in Figure 1, despite the variation in pre- and post-test probabilities among studies, differences between these probabilities within a study were not as variable, with only 12 of 50 (24%) combinations of population × test × result showing upward or downward effects of more than one category. With strict observance of 95% CIs, this fraction decreased to 4 of 50 (8%).
Simplified application of Bayes theorem to evidence-based diagnosis
Medow and Lucey41 suggest a simplified approach to the use of Bayes theorem to estimate posttest diagnostic probabilities. The authors note that, for the many tests with sensitivity and specificity in the range 0.80–0.90 (e.g., cerebrospinal fluid protein tests for sporadic Creutzfeldt–Jakob disease; Appendix 1), applying Bayes theorem typically modifies diagnostic probability upward or downward by one step among five different heuristic, general-purpose probability categories: very unlikely (< 0.10), unlikely (0.10–0.33), uncertain (0.33–0.67), likely (0.67–0.90) and very likely (> 0.90). For example, if the clinician’s initial diagnostic opinion is unlikely or very unlikely, positive results for many commonly ordered tests when used alone are not powerful enough to revise the posttest probability to the very likely or likely category. The converse applies when pretest diagnostic probability is high and a negative test result is returned. In practice, a judgment of “very unlikely” may, for example, prompt a decision to prioritize other diagnostic hypotheses; “very likely” may elicit a decision to proceed with treatment; and “uncertain” will usually indicate a need for further testing. Other definitions of qualitative diagnostic probability categories are of course possible (e.g., when decisions require different action thresholds, or test performance characteristics fall < 0.80 or > 0.90), but Medow and Lucey argue that the division described above tends to reflect well the decision thresholds used in practice by many clinicians for diverse conditions and supporting investigations.
In some studies, more than one test was performed on the same patients, which enabled direct comparisons of accuracy between markers (Figure 1). In nearly all such cases, tau modified diagnostic probabilities more strongly than 14-3-3, particularly with positive test results. Moreover, in five studies, a positive result with 14-3-3 did not significantly shift diagnostic probability to the neighbouring category, whereas all studies of tau showed significant shifts of at least one category. In three studies, it was feasible to directly compare the performance of S100B with that of the other two markers. In two of these, the accuracies of S100B and tau were closely comparable.
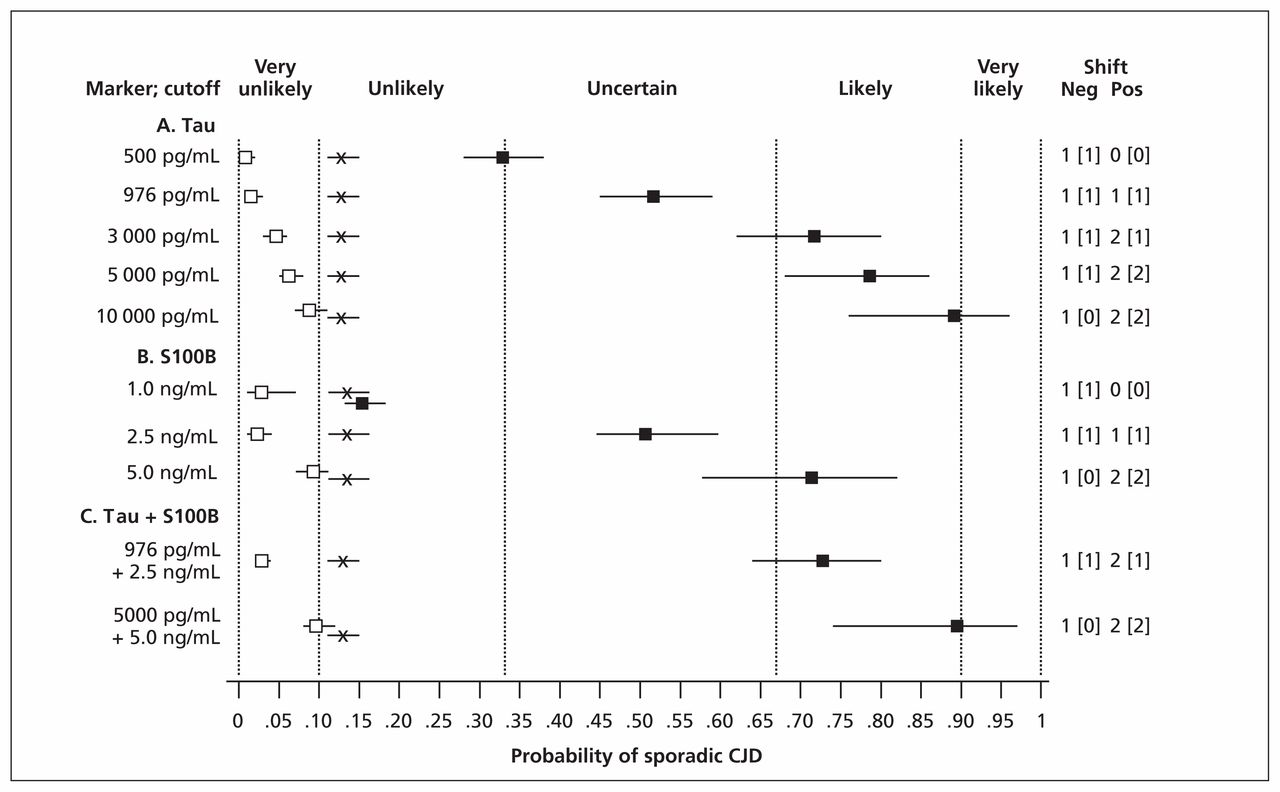
Using the data of Coulthart and colleagues15 for tau and S100B, we also examined the effects of using different test-scoring thresholds and of combining the two markers. As shown in Figure 2, both of these approaches increased discriminatory power, particularly with positive test results. For example, a tau concentration greater than 976 pg/mL (the optimal intermediate threshold in the study) modified a pretest probability of 0.13, or unlikely, to a posttest probability of 0.52 (95% CI 0.45–0.58), or uncertain. At a concentration greater than 10 000 pg/mL, which was observed in 41 of 120 patients with sporadic CJD,15 posttest probability became 0.89 (95% CI 0.79–0.96), or likely. When tau and S100B results were combined, posttest probabilities increased from 0.52 (95% CI 0.45–0.58) and 0.50 (95% CI 0.44–0.59), both uncertain, at individual intermediate thresholds of 976 pg/mL and 2.5 ng/mL, respectively, to 0.73 (95% CI 0.64–0.80), or likely, at the same thresholds. At still higher thresholds of 5000 pg/mL and 5.0 ng/mL, respectively, a combined positive result yielded a posttest probability of 0.89 (95% CI 0.74–0.97), again likely, but verging on very likely.
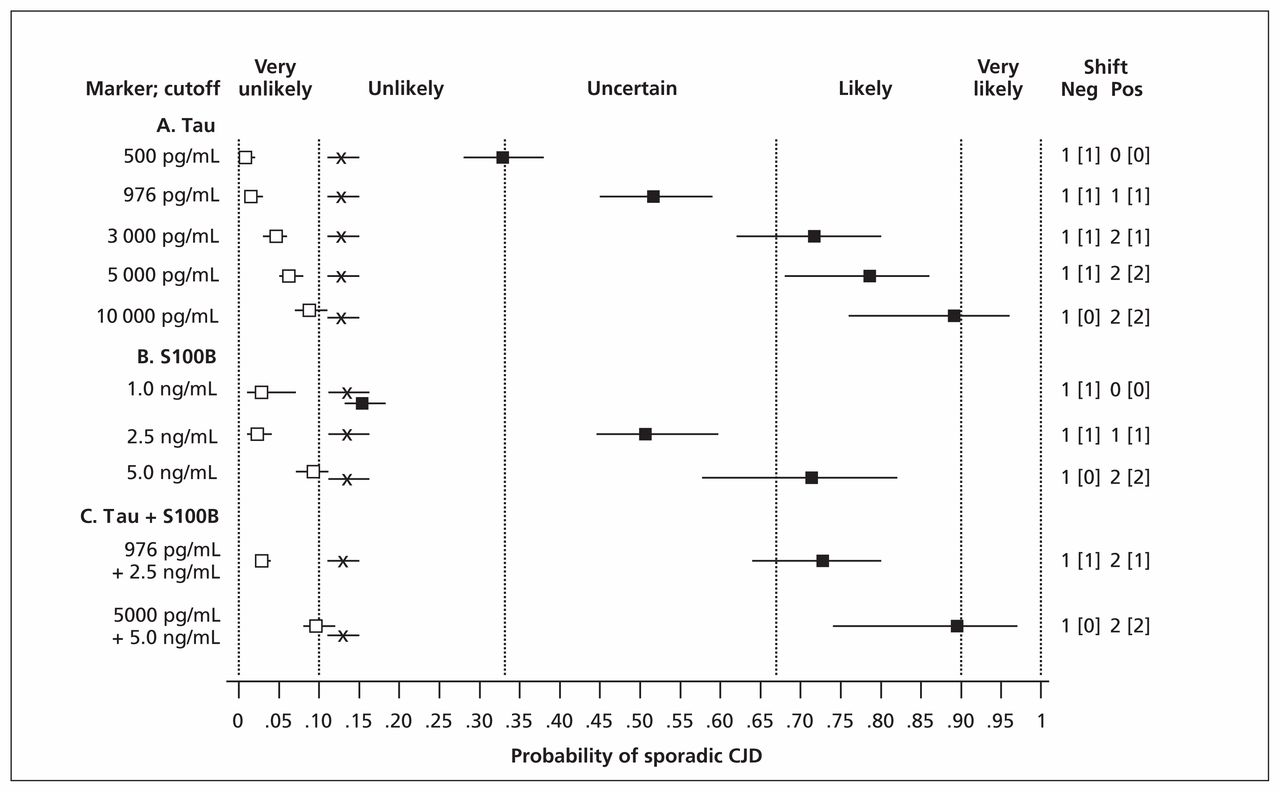
Pre- and posttest diagnostic probabilities for sporadic Creutzfeldt–Jakob disease (CJD), based on the data of Coulthart and colleagues,15 for various illustrative concentrations of cerebrospinal fluid tau and S100B protein individually and in combination. X: pretest probability (sporadic CJD prevalence); closed and open squares: posttest probability for positive or negative test results, respectively; horizontal bars: 95% confidence interval (CI); vertical dashed lines: diagnostic opinion category (Box 2); numbers at right: size of effect on diagnostic probabilities in terms of category differences, for negative (Neg) and positive (Pos) test results; numbers in square brackets: effect size with strict application of 95% CI.
How can an evidence-based approach guide clinical practice?
We placed published data about three cerebrospinal fluid protein markers (14-3-3, tau and S100B) within an evidence-based framework to clarify the interpretation of test results in diagnostic investigations of sporadic CJD. We observed wide variation among studies in pre- and posttest probabilities, and some variation in test performance. However, the effects of these tests on diagnostic probabilities are fairly uniform, with statistically significant shifts between at most two probability categories among those proposed by Medow and Lucey (Box 3).41 For tau and S100B, varying scoring thresholds and combining test results can enhance test power. Thus, although caution remains warranted, we suggest that the utility of cerebrospinal fluid protein tests need not be restricted to clinical settings in which pretest probability of sporadic CJD has been highly refined by prior investigations. To illustrate how these insights might be applied in practice, two fictional cases are presented (Box 4).
Fictional cases
Case 1: A 67-year-old retired teacher presented to her family physician with recent memory problems. On examination, her Montreal Cognitive Assessment score was 25/30, and there was a subtle dysdiadochokinesis of the right upper extremity. When seen by a neurologist three weeks later, she had frank ataxia and was unable to follow commands. There was no family history of similar conditions. On admission to the regional hospital, electroencephalography (EEG) and magnetic resonance imaging (MRI) of the brain were ordered, lumbar puncture performed and a range of tests ordered. Within days the patient’s condition had progressed dramatically: she was bedridden, had startle myoclonus and Babinski reflexes, and barely communicated. At this point the neurologist’s diagnostic opinion was that sporadic Creutzfeldt–Jakob disease (CJD) was likely (70%–80% probability). Cerebrospinal fluid results were returned as positive for 14-3-3 protein, with tau protein greater than 12 000 pg/mL and S100B protein greater than 4 ng/mL, indicating a posttest probability of very likely (> 90%) for sporadic CJD. Meanwhile, MRI results had shown hyperintensity in the putamen and caudate nucleus head, and EEG showed periodic sharp waves at about 1 Hz, both also consistent with sporadic CJD. Because all other investigations proved uninformative, a final clinical diagnosis of probable sporadic CJD was shared with the patient’s family, consent for autopsy was requested and granted, and palliative care was arranged. The patient died 12 weeks after onset of symptoms. On autopsy, the brain showed the pathologic hallmarks of CJD.
Case 2: A 58-year-old widower with a recent history of heavy drinking and poor nutrition had experienced mild dizziness and unsteadiness of gait for several months when his son noticed that he had become forgetful and was having difficulty walking. The forgetfulness progressed rapidly in the next several weeks, until one morning he awoke in a confused state, had difficulty speaking and was unable to perform simple routine tasks. He was taken to the emergency department at a local hospital, where he walked with an ataxic gait. On examination he exhibited loss of short-term memory, and his Montreal Cognitive Assessment score was 19/30. There was mild sensory loss in the sock region bilaterally, an essential tremor of both hands and moderate disturbance of the heel–shin test bilaterally. His son also mentioned that on several occasions he had exhibited “jerks” in his right hand and arm. However, myoclonus was not observed. Because this patient had rapidly progressing dementia, ataxia and possibly myoclonus, the hospital’s consulting neurologist raised the possibility of sporadic CJD, although he considered it very unlikely (< 10%). A lumbar puncture was performed, and a range of tests ordered including cerebrospinal fluid protein markers for sporadic CJD. Because alcohol-related Wernicke encephalopathy was also suspected, a blood sample was sent out for thiamine analysis. Meanwhile, the patient was admitted and empirical treatment started with infusions of thiamine hydrochloride. Cerebrospinal fluid marker results were returned as negative for 14-3-3, with tau 593 pg/mL and S100B 0.9 ng/mL, indicating a posttest probability for sporadic CJD of very unlikely (< 1%). The thiamine result was 28 nmol/L (laboratory-specific reference range, 70–180 nmol/L), supporting the diagnosis of Wernicke encephalopathy. Because the patient had begun to respond to thiamine treatment, it was continued, and within a month he had made a full recovery.
A well-defined pretest diagnostic opinion is also essential to derive maximum benefit from any test. Approached informally, this process can be unreliable and even misleading.29,42 However, the clinician can sometimes draw on population-based estimates of prevalence for a target condition in relevant clinical settings.43 An estimate of this kind is available from a prospective sample of 1000 Canadian patients for whom a suspicion of sporadic CJD prompted cerebrospinal fluid testing, and in whom the prevalence of autopsy-confirmed sporadic CJD was about 13%.15 Additional prevalence estimates for sporadic CJD would prove useful, particularly if based on prospective samples of adequate size and accompanied by detailed descriptions of clinical setting. The clinician’s view of pretest probability may also be incorporated into an evidence-based framework with formal approaches, perhaps with a population-based prevalence estimate as a starting point for refinement of expert opinion.44 To take account of statistical uncertainty, interval estimates (e.g., 95% CIs) for pre- and posttest probabilities should also be kept in mind. This is particularly important for any rare disease, for which study populations are necessarily smaller and CIs wider.44
Apart from guiding thinking about a specific diagnosis, a probability-based framework can facilitate other clinical decisions. As mentioned (Box 3), intermediate probability values (e.g., 0.1–0.9) typically indicate a need for further investigation, whereas higher or lower values (e.g., < 0.1 or > 0.9) may trigger treatment or intensified investigation of alternative diagnoses.41,45 Although therapies are not yet available for sporadic CJD, other important decisions include whether, when and how to communicate with a patient’s family about the diagnosis, arrange palliative care, request autopsy or undertake potentially complex or costly investigations for non-CJD conditions that may be treatable (e.g., infections, autoimmune diseases and toxicologic reactions). Some decisions, however, (e.g., those related to infection control) may require diagnostic probability thresholds more stringent than those currently attainable in the living patient, and thus will be guided primarily by precaution.46 Definitive laboratory diagnosis of human prion diseases in living patients may become possible with new approaches such as ultrasensitive detection of PrPSc in body fluids or use of novel protein markers.17,47 However, even when such approaches become available, a structured, evidence-based approach will play a pivotal role in their evaluation and application.
Key Points-
Prion diseases are rare degenerative brain disorders that are challenging to diagnose yet potentially transmissible by infection; the most common type is sporadic Creutzfeldt–Jakob disease (CJD).
-
Definitive diagnosis of prion diseases requires neuropathologic examination (usually postmortem), but clinical, paraclinical and laboratory criteria can be useful in diagnostic assessment.
-
For sporadic CJD, diagnostic accuracy has been characterized for laboratory assays of several proteins found in cerebrospinal fluid.
-
Given an appropriate evidence-based framework, information from laboratory tests can be useful in refining the probability of a diagnosis of sporadic CJD.
Acknowledgements
We extend our gratitude to Canadian patients, families, physicians and other collaborators of the Canadian Creutzfeldt–Jakob Disease Surveillance System (CJDSS) for their generous contributions to surveillance and study of human prion diseases in Canada, and to all members of the CJDSS team for their dedication and support. We are also grateful to Bernard Choi, Zheng Wang, Nicholas Ogden and Michel Deilgat for providing helpful comments on an earlier version of the manuscript, and to Byron Wallace for assistance with OpenMeta[Analyst].
Footnotes
-
Competing interests: None declared.
-
This article has been peer reviewed.
-
Contributors: Michael Coulthart conceived the idea for the article and conducted the literature review. All of the authors drafted and revised the article, and approved the version submitted for publication.
References
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

